Wie Morbus Fabry und Morbus Gaucher gehört auch Morbus Hunter (MH) mit einer Prävalenz von bis zu ca. 1:140.000 weltweit zu einer speziellen Klasse der lysosomalen Speicherkrankheiten (LSD), den Mukopolysaccharidosen (MPS), und wird auch als MPS II bezeichnet.1
MH folgt einem X-chromosomal rezessiven Erbgang. Aus diesem Grund sind fast ausschließlich Männer betroffen.2
Grundsätzlich werden zwei Verlaufsformen unterschieden: eine nicht-neuronopathische Form, die ca. 25 % der Patienten betrifft, und eine neuronopathische Verlaufsform.
Erste neuronopathische Symptome können sich meist ab einem Alter von 18 Monaten manifestieren, während nicht-neuronopathische Symptome in der Regel zwischen dem 2. und 4. Lebensjahr auftreten.1,3,4 Unbehandelte Patienten mit einer neuronopathischen Verlaufsform versterben meist im frühen Erwachsenalter an Herzversagen oder obstruktiven Lungenerkrankungen. Bei Patienten mit nicht-neuronopathischer Verlaufsform beträgt die Lebenserwartung in der Regel zwischen 50 und 60 Jahren.1,5,6
Ursache von MH ist eine Mutation des IDS-Gens, das für das lysosomale Enzym Iduronat-2-Sulfatase (I2S) kodiert.2 Unter physiologischen Bedingungen ist I2S für den Abbau von bestimmten Glykosaminoglykanen, spezifisch Dermatansulfat und Heparansulfat, zuständig. Diese sind u.a. Bestandteile stabilisierender Strukturen in der Zellmembran und der extrazellulären Matrix.7 Bei MH ist der Abbau dieser Stoffe gestört - sie sammeln sich im Lysosom an, wodurch die physiologischen Funktionen der Zelle gestört werden.2 Diese Störung führt zu einer Vielzahl von Symptomen.2 Bislang sind mehr als 300 verschiedene Genvarianten bekannt, die die Proteinexpression, -funktion oder den lysosomalen Transport von I2S beeinträchtigen.2
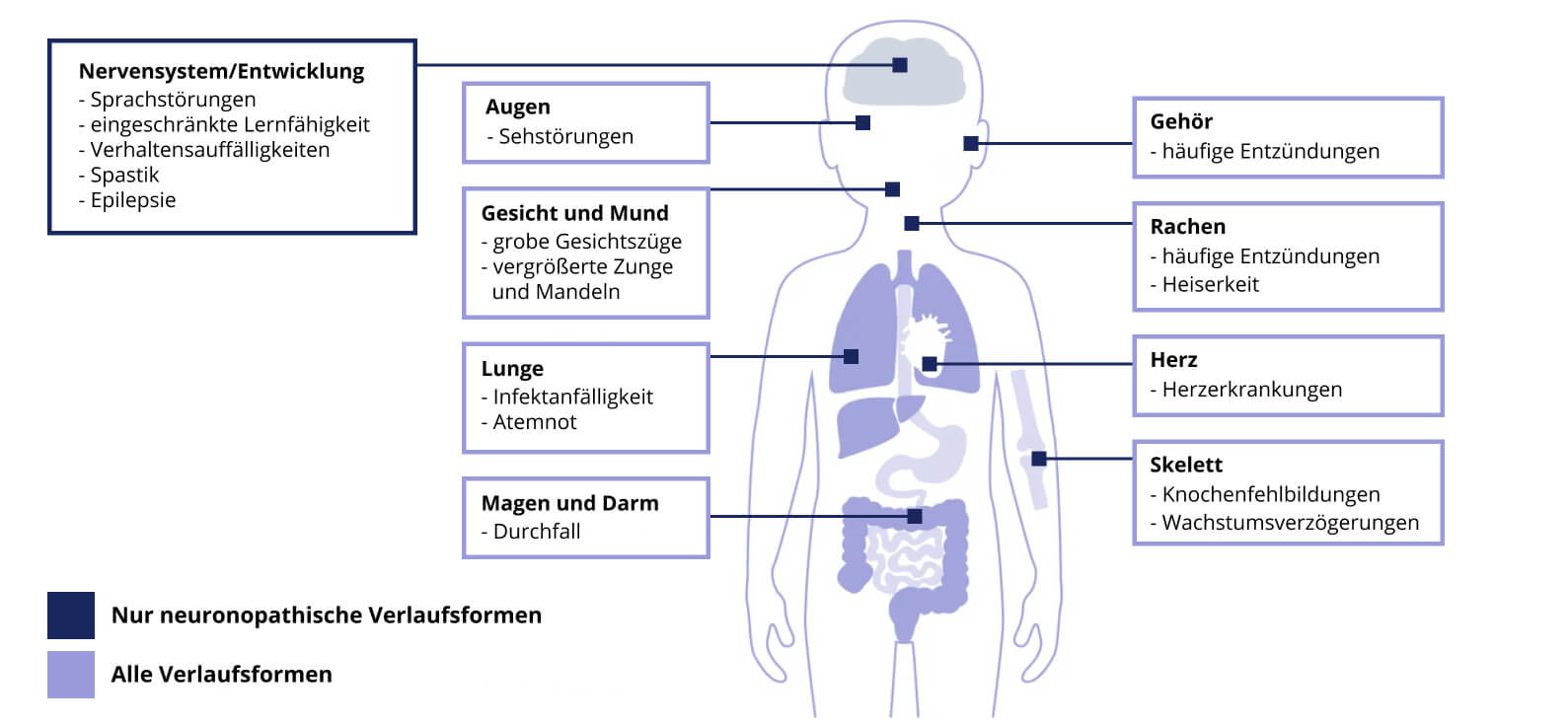
MH ist eine multisystemische Erkrankung mit einem variablen und heterogenen Verlauf (Abb.1). In der Regel entwickeln sich zusätzliche Symptome im Laufe des Lebens bei gleichzeitigem Fortschreiten der Schwere bestehender Symptome.1,5,6
Abb. 1: Übersicht der möglichen Symptome von Morbus Hunter.
Da sich viele der Symptome mit üblichen Kindheitsbeschwerden überschneiden, bleibt Morbus Hunter häufig unerkannt.1 Wenn sich MH-assoziierte Symptome häufen, ist eine tiefergehende Anamnese des Patienten empfehlenswert.1
MH kann durch verschiedene Laboruntersuchungen nachgewiesen werden. Zum einen kann mithilfe eines Urintests die Konzentration von Dermatan- und Heparansulfat gemessen werden.2 Ein erhöhter Wert gibt Anzeichen dafür, dass die I2S-Funktion eingeschränkt ist. Eine weitere Möglichkeit stellt die Messung der I2S-Aktivität im Blut mittels eines Trockenbluttests dar.
Zusätzlich kann MH pränatal nachgewiesen werden, denn die I2S-Aktivität ist auch im Plazentagewebe oder Fruchtwasser messbar .2 Darüber hinaus ist die Messung der Dermatan- und Heparansulfatkonzentration im Fruchtwasser möglich.2 Pränatale Untersuchungen empfehlen sich allerdings nur, sofern eine familiäre Vorbelastung bekannt ist.2
Letztendlich muss die Diagnose mithilfe einer molekulargenetischen Untersuchung des IDS-Gens gesichert und die genaue vorliegende Mutation identifiziert werden.2 Die Diagnostik von MH findet meist in spezialisierten Zentren oder über Diagnostikinitiativen statt. Bei Letzteren können auch Tests für die Verwendung in der Praxis bestellt werden.
Die Behandlung von MH sollte idealerweise in spezialisierten Stoffwechselzentren begonnen werden. Dabei wird eine Enzymersatztherapie (EET) verwendet, um den Enzymmangel auszugleichen.
Zusätzlich wird derzeit an weiteren Therapiemöglichkeiten geforscht, wie bspw. Stammzelltherapien.5,10
Um die vielfältigen Symptome des MH zu behandeln, sind abhängig vom jeweiligen Krankheitsbild diverse Begleittherapien nötig. Daran sind eine Vielzahl von Spezialist:innen wie u.a. Kardiolog:innen, Orthopäd:innen, Neurolog:innen, Pneumolog:innen und HNO-Ärzt:innen beteiligt.
Falls sie noch weitere offene Fragen zum Thema Morbus Hunter haben, finden Sie hier zahlreiche zusätzliche Informationen.
EXA/DE/FAB/0276_09/2024