Morbus Gaucher (MG) gehört, wie auch Morbus Fabry (MF), mit einer Inzidenz von 1:40.000 bis 1:60.000 zu den seltenen lysosomalen Speicherkrankheiten.1 Auch MG ist eine Erbkrankheit und wird autosomal rezessiv vererbt.1 Daher können sowohl Männer als auch Frauen betroffen sein.
Da verschiedene Organsysteme betroffen sind, zählt MG zu den Multiorganerkrankungen. Die Symptome sind höchst heterogen, ebenso wie ihre Schweregrade.1 Je nach Ausprägung des MG können diese bereits im frühen Kindesalter auftreten, allerdings ist auch eine Manifestation im Erwachsenenalter möglich.1 Eine Manifestation bei Patient:innen jünger als 5 Jahre korreliert dabei mit einem höheren Risiko für einen schweren Krankheitsverlauf.2
Die häufigste Ursache von MG ist eine Mutation des Glukozerebrosidase-Gens (GBA), das für das lysosomale Enzym beta-Glukozerebrosidase kodiert.1 In gesunden Lysosomen ist beta-Glukozerebrosidase für den Abbau der Lipidsubstanz Glukozerebrosid verantwortlich. Diese sind ein wichtiger Bestandteil der Zellmembran.1,2 Ist die Funktion des Enzyms eingeschränkt oder nicht mehr vorhanden, reichern sich die Glukozerebroside in den Lysosomen an.1 Das geschieht hauptsächlich in Makrophagen. Betroffene Zellen lassen sich leicht unter dem Mikroskop anhand ihrer aufgedunsenen Lysosomen erkennen und werden auch Gaucher-Zellen genannt. Diese Gaucher-Zellen sind besonders in Milz, Leber, Knochenmark und der Lunge zu finden.
Bislang sind mehr als 400 verschiedene Genvarianten bekannt, die die normale Funktion der beta-Glukozerebrosidase einschränken.1
In sehr seltenen Fällen kann anstatt der GBA-Mutation eine Mutation im Prosaposin-Gen (PSAP) die Ursache für MG sein.1
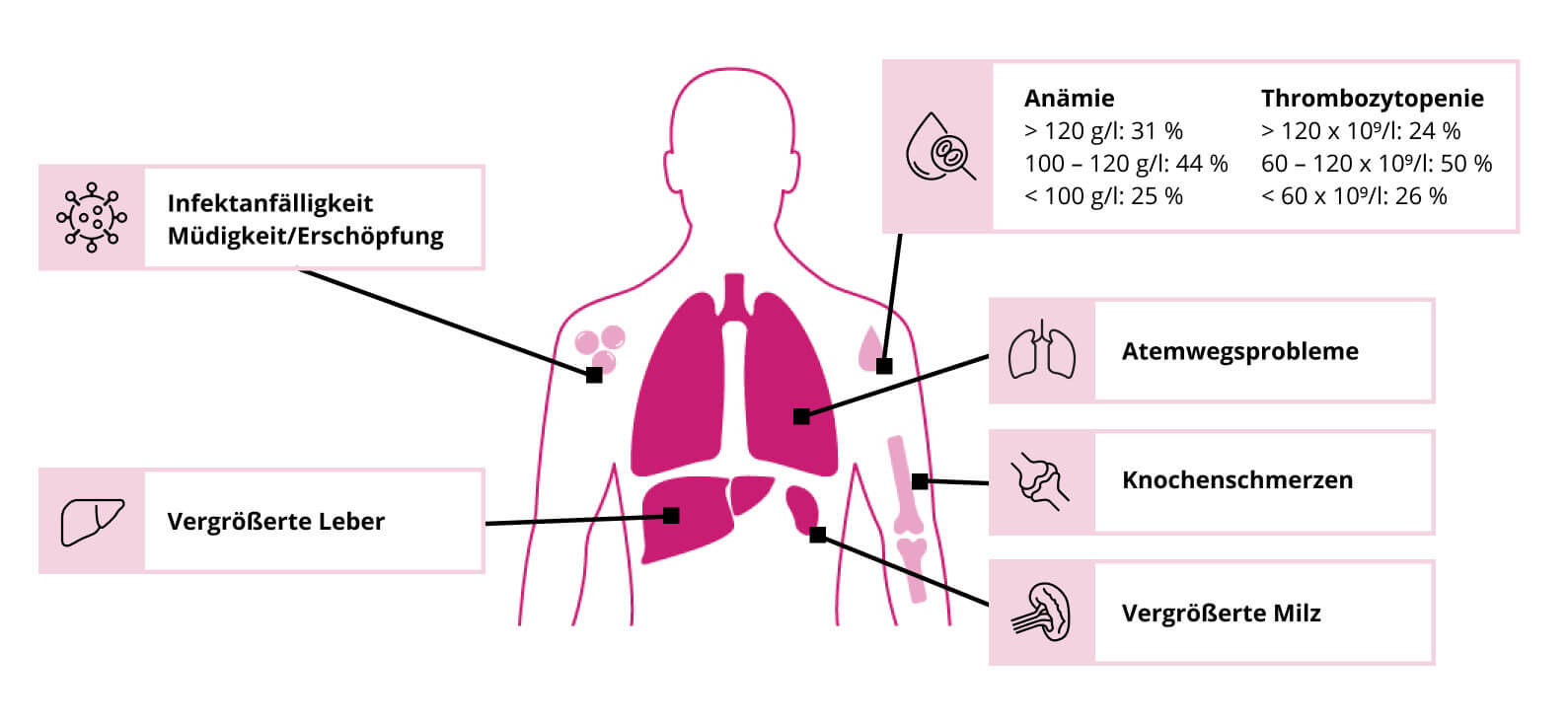
Je nach Manifestationsalter und Vorliegen neuronaler Symptome werden drei Formen von MG mit unterschiedlichen Prognosen unterschieden: Typ I, Typ II und Typ III. Typ I ist die häufigste und mildeste Verlaufsform. Sie betrifft ca. 90-95 % aller Patient:innen mit MG und weist häufig eine Kombination aus verschiedenen viszeralen, hämatologischen und ossären Symptomen auf (Abb.1).1 Typ II und Typ III sind im Vergleich zu Typ I schwerere Verlaufsformen und gekennzeichnet durch zusätzliche neurologische Symptome.
Abb. 1: Übersicht häufiger Symptome und betroffenere Organe bei Typ I Morbus Gaucher. Bei der Anämie wurde die Häufigkeit der einzelnen Hämoglobinkonzentrationen angeben, während bei der Thrombozytopenie die Häufigkeit die Thrombozytenzahl pro Liter angeben wurde. Modifiziert nach 3
Wie bereits erwähnt, sind die Symptome des MG hoch heterogen. Deshalb sollte bei Symptomatiken, die viel häufigere Erkrankungen wie bspw. Leukämie, Herzinsuffizienz oder das multiple Myolom vermuten lassen, auch an MG gedacht werden.,4
Bei Verdacht auf MG kann die Aktivität der beta-Glukozerebrosidase durch eine Blutuntersuchung, wie bspw. einen Trockenbluttest, gemessen werden.1 Zusätzlich kann das Vorliegen bestimmter Biomarker im Blut überprüft werden.1 Zu diesen zählen u.a. CLL-18, das direkt von Gaucher-Zellen sezerniert wird, und Lyso-GB1, ein Glycosylsphingosin, das bei gestörtem Abbau von Glukozerebrosid entsteht.1
Zudem kann eine molekulargenetische Analyse des GBA-Gens die Diagnose sichern und die vorliegende Genvariante identifizieren.1 Tests können bei spezialisierten Diagnostikinitiativen angefordert werden.
Neben der Diagnose der zugrundeliegenden Erkrankung ist es ebenso wichtig, den Schweregrad der Krankheit zu identifizieren. Dafür werden Ultraschalluntersuchungen und andere bildgebende Verfahren benutzt, um die Knochen- und viszerale Beteiligung zu bestimmen.1
Genaue Therapieeinstellungen sollten in spezialisierten Morbus Gaucher Kompetenzzentren vorgenommen werden.
Derzeit sind zwei kausale Therapiestrategien für die Behandlung des MG zugelassen - die Substratreduktionstherapie (SRT) und die Enzymersatztherapie (EET).1,5,6 Um den Leidensdruck der Patient:innen zu reduzieren, sind auch symptomatische Behandlungen nötig. Infolgedessen werden Patient:innen oft von einem interdisziplinären Team betreut.1,5,6
Benötigen Sie zusätzliche Informationen rund um das Thema Morbus Gaucher? Dann klicken sie hier.
EXA/DE/FAB/0276_09/2024