Zum ersten Mal haben Forscher mit Hilfe einer Genschere Patienten mit schweren Blutkrankheiten behandelt. Ob die Gentherapie es nun in die Praxis schafft? Eine Einschätzung.
„Wir stehen am Anfang von etwas ganz Großem“, so Prof. Dr. Toni Cathomen von der Uniklinik Freiburg über die „Genschere“ CRISPR/Cas9. Mit dem Verfahren soll es Forschern aus Regensburg und aus Nashville, Tennessee, erstmals gelungen sein, zwei Patienten mit Beta-Thalassämie erfolgreich zu behandeln. DocCheck berichtete. Kommt jetzt, nach etlichen Misserfolgen und wenigen Zulassungen, neuer Schwung in die Gentherapie?
Ursprünglich stammt das CRISPR/Cas-System aus Bakterien, die sie vor viralen Infektionen schützen. Das Erbgut des Eindringlings wird erkannt und gezielt geschnitten. Doch warum gilt die „Genschere“ bei Medizinern als große Innovation? Das liegt vor allem an der außergewöhnlichen Präzision der Genschere. Das Protein CAS9 trägt eine kurze Basen-Sequenz mit sich, um eine bestimmte Region im Erbgut anzusteuern und zu schneiden. Durch das Einsetzen neuer Sequenzen an Schnittstellen lassen sich Gene komplett austauschen – etwa eine mutierte, inaktive Form durch die natürliche Variante.
CRISPR/Cas9 ist nicht die einzige Methode, um Gene zu manipulieren. Das Verfahren gilt aber als präziser, preisgünstiger und schneller als ähnliche Technologien. Es könnte 30 Jahre nach ersten, desaströsen Experimenten der Gentechnologie neuen Schwung verleihen.
Doch noch einmal zurück zum Start: Alles begann mit Verbesserungen bei der Technik der Genentschlüsselung und mit steigenden Computerleistungen. Zwischen 1990 und 2001 sequenzierten 1.000 Wissenschaftler aus 40 Ländern das menschliche Erbgut. Gleichzeitig explodierte die Zahl genomweiter Assoziationsstudien (GWAS). Hier werden Assoziationen genetischer Varianten in Bevölkerungsgruppen mit Phänotypen, beispielsweise Krankheiten, untersucht. Plötzlich erkannten Humangenetiker, welche Mutationen in Genen für ein bestimmtes Leiden ausschlaggebend sind. Die Datenbank orpha.net nennt insgesamt 6.172 seltene Erkrankungen, von denen 71,9 Prozent genetischen Ursprungs sind. Was lag näher, als defekte Teile im Erbgut durch die richtige Sequenz zu ersetzen?
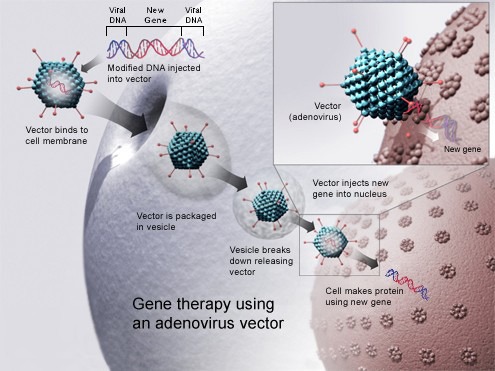
Gesagt, getan. Bereits 1972 äußerten Forscher – wenn auch nur theoretisch – die Hoffnung, fehlerhafte Gene im Erbgut künftig austauschen zu können. Und in 1984 wurde das erste Transportvehikel für Gene vorgestellt: ein Vektor auf Basis des Simian-Virus 40. Damit lassen sich veränderte Gene in den menschlichen Körper einschleusen.
Gentherapie mit einem viralen Vektor. Grafik: NIH/Wikipedia, CCO
Im Jahr 1990 ging es dann richtig los: Trotz etwaiger Bedenken erhielt eine vierjährige Patientin mit schwerem kombinierten Immundefekt (SCID) die weltweit erste experimentelle Gentherapie. Schon ein grippaler Infekt kann bei SCID zum Tode führen. Ärzte entnahmen der Patientin unterschiedliche Leukozyten, modifizierten diese gentechnologisch und brachten sie zurück in den Blutkreislauf. Das Verfahren funktionierte, wenn auch nur für mehrere Monate. Dann mussten weitere modifizierte Leukozyten verabreicht werden.
Bei der X-SCID, der häufigsten Form einer SCID, ist die Mutation bekannt, was gezielte Therapien ermöglicht. Zwischen 1999 und 2000 behandelten Forscher fünf an X-SCID erkrankte Kinder. Als Vektor verwendeten sie das Murine Leukämievirus (MLV). Bei vier von fünf kleinen Patienten glückte die Gentherapie, und ihr Immunsystem normalisierte sich. Weitere Behandlungen folgten, bis man 2003 feststellte, dass bei zwei von insgesamt zehn Patienten Leukämien aufgetreten waren. Das lag vermutlich am MLV selbst; es hatte sich nahe einer Sequenz ins Erbgut eingebaut, welche Krebs fördert.
Damit nicht genug: Im Jahr 1999 starb ein Patient im Zuge der Gentherapie an Multiorganversagen durch die virale Infektion. Er wurde zuvor aufgrund seiner angeborenen Stoffwechselerkrankung, eines Ornithin-Transcarbamylase-Defizits, mit genetisch veränderten Adenoviren behandelt. Kurz darauf stoppte die US Food and Drug Administration (FDA) alle weiteren Experimente.
Das Desaster warf Forscher um Jahre zurück – sie untersuchten Gentherapien nur noch in Tiermodellen. Schließlich wagte man sich wieder an klinische Studien der Phase 1, unter anderem bei Patienten mit X-chromosomaler chronischer Granulomatose (X-CGD), einer angeborenen Immunschwäche (2006) oder bei der Leberschen Congenitalen Amaurose (2008), einer genetisch bedingen Sehbeeinträchtigung. Besonders häufig wurden genetisch ausgelöste Stoffwechselerkrankungen in dieser Phase untersucht. Bei der familiären Lipoproteinlipasedefizienz (LPLD) gelang es uniQure in 2012 sogar, Glybera® als weltweit erstes Gentherapeutikum zuzulassen. In 2017 hat uniQure die Erlaubnis nicht verlängert, wahrscheinlich aus wirtschaftlichen Gründen.
Zur Behandlung der anfangs erwähnten Beta-Thalassämie wurde 2019 Zynteglo® zugelassen. Bei der Herstellung modifizieren Ärzte aus dem Blut ihres Patienten entnommenen Stammzellen durch ein Virus, welches Arbeitskopien des Beta-Globin-Gens in die Zellen überträgt. Wenn diese modifizierten Zellen nach einer Chemotherapie reinfundiert werden, gelangen sie zum Knochenmark. Dort bilden sie funktionsfähige Erythrozyten. Doch die Therapie hat ihren Preis: 1,5 Millionen Euro pro Einzeldosis.
In den USA macht derzeit Zolgensma® von Novartis Schlagzeilen – ebenfalls aufgrund des hohen Preises. Das Gentherapeutikum soll die Spinaler Muskelatrophie heilen, einer angeborenen Erkrankung mit Muskelschwäche und Lähmung der Atemmuskulatur. Die FDA hat ihren Segen erteilt. Nur können sich die meisten amerikanischen Eltern umgerechnet 1,9 Millionen Euro pro Spritze nicht leisten. Eine EMA-Zulassung steht noch aus. Hinzu kommt: Weitere Studien mit Zolgensma® wurden von der FDA aufgrund von Sicherheitsbedenken kürzlich auf Eis gelegt.
Bei Gentherapien geht es aber nicht nur um angeborene Erkrankungen. Mit den CAR-T-Zell-Therapien zeigen Forscher den Nutzen auch bei Krebs. CAR steht für Chimeric Antigen Receptor. T-Zellen werden aus dem Blut von Patienten gewonnen und im Labor gentechnisch „scharf gemacht“. Sie bilden dann chimäre Antigenrezeptoren (CAR) auf ihrer Oberfläche, die sich gegen Oberflächenproteine von Krebszellen richten.
Trotz einiger Zulassungen sei gesagt: Nach 30-jähriger Forschung zu Gentherapien ist das Ergebnis alles in allem recht mager. Vielleicht bringt CRISPR/Cas9 jetzt den lang erwarteten Durchbruch. Doch selbst das hoch gelobte Verfahren hat Schwächen: Beide Stränge werden geschnitten, was bei der Reparatur auch zu unerwünschten Ergebnissen führen kann.
Wie sich diese Schwäche umgehen lässt, zeigen Forscher vom Broad Institute in Cambridge, Massachusetts. Beim sogenannten Prime Editing arbeiten sie mit zwei modifizierten Proteinen und einer spezifischen RNA-Vorlage, um Ziele im Erbgut zu erkennen. Ihr System kann einzelne DNA-Bausteine austauschen, entfernen oder neu kombinieren – ohne unerwünschten Schnitt der Doppelhelix. Es soll noch präziser als CRISPR/Cas9 arbeiten, zumindest im Experiment.
Bislang führten die Forscher mit ihrem Prime Editing 175 Veränderungen in menschlichen Zelllinien sowie in Nervenzellen von Mäusen durch. Beispielsweise korrigierten sie die Sichelzellanämie und dem Tay-Sachs-Syndrom (einer Stoffwechselstörung aufgrund des fehlenden Enzyms β-N-Acetylhexosaminidase) in vitro. Wann Forscher zum Sprung in die Praxis ansetzen, ist nicht bekannt.
Bildquelle: Jude Beck, unsplash