Normalmente, cuando alguien afirma que una enfermedad es apasionante, suele querer decir que no hay quien la entienda. Las queratodermias palmo-plantares hereditarias son, pues, apasionantes. Y mi misión a lo largo de esta entrada es aportar algo de luz a este complejo mundo que ni yo comprendo del todo. Agarraos que vienen curvas. Y genes, muchos genes.
Y es que las queratodermias palmo-plantares (QPP) son un grupo heterogéneo de enfermedades que se caracterizan por un engrosamiento anómalo de la piel de palmas y plantas, cuyo correcto diagnóstico es a menudo complicado por muchos motivos. En primer lugar, tendremos que diferenciar las QPP hereditarias de las adquiridas, estas últimas más frecuentes y habitualmente de aparición más tardía, y de variada etiología: queratodermia climatérica, psoriasis, dermatitis de contacto, malnutrición, fármacos, químicos, neoplasias, infecciones y otras dermatosis. Pero lo que hoy nos ocupa son las QPP hereditarias, otro gran grupo de enfermedades de difícil diagnóstico por su gran heterogeneidad clínica y genética y por la probable existencia de síndromes aún no identificados.
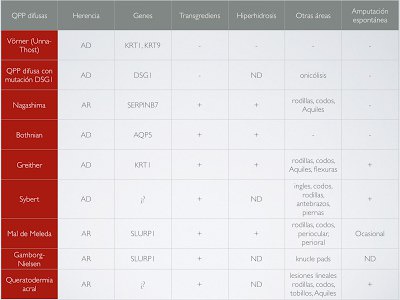
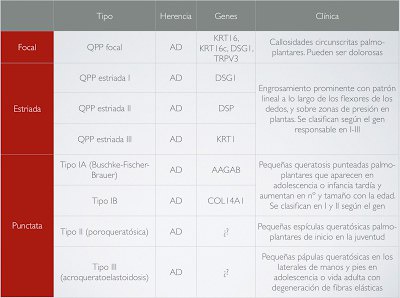
En general, las QPP hereditarias se dividen en dos grandes grupos: aquellas sin otros hallazgos asociados y las QPP con hallazgos asociados (nos referimos a afectación de otras estructuras, como las uñas, el pelo, dientes e incluso la afectación de otros órganos). Pero morfológicamente existe otra clasificación que las divide en 4 tipos: difusa, focal, estriada y punctata. Dentro de cada tipo van encajando las diferentes QPP descritas hasta el momento, como resumen las tablas 1 y 2 que ilustran esta entrada, según el reciente artículo de Sakiyama (J Dermatol. 2016;43:264). No voy a entrar en detalles de todas las variantes porque podríamos estar horas (y así dejamos algo de material para otro día), pero sí os comentaré una palabreja que veréis escrita más de una vez cuando hablamos de las QPP difusas: transgrediens. Es un hallazgo clave en la aproximación diagnóstica de estas enfermedades y que hace referencia a la expansión de las lesiones a las superficies dorsales de manos y pies, muñecas y región aquílea. El mal de Meleda es un buen ejemplo de QPP difusa con transgrediens.
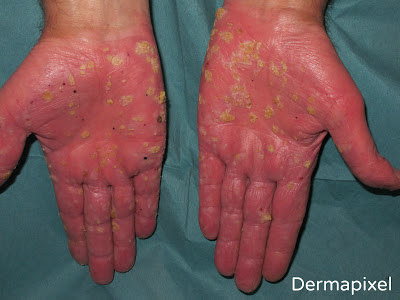
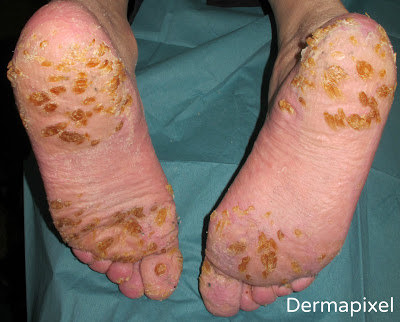
Pero volvamos a Fermín, nuestro paciente de esta semana. Un paciente con este tipo de lesiones que habían ido empeorando desde la adolescencia y con antecedentes familiares (siguendo un patrón autosómico dominante) hacía pensar en algún tipo de QPP de tipo punctata, probablemente de tipo I por la clínica que presentaba. Lo cierto es que, dadas las circunstancias, no realizamos más pruebas al pobre Fermín, que lo único que quería era que sus calcetines aguantaran un poco más, y a quien poco le importaba si lo que tenía era una QPP punctata tipo IA o IB (sólo lo podríamos haber sabido con estudios genéticos que no se realizan rutinariamente). La QPP de Buschke-Fischer-Brauer fue descrita por primera vez en 1879 como “clavos diseminados de manos y pies”. En 1910, Buschke y Fisher la documentaron mejor y en 1913 Brauer demostró su naturaleza hereditaria. No fue hasta 2005 cuando un estudio chino evidenció una región en el cromosoma 15q22.2-15q22.31 como el locus genético de este síndrome. Con una incidencia estimada de 1.17/100.000, empieza a manifestarse al final de la infancia o adolescencia, aunque existen casos reportados de inicio en la 5ª década de la vida. Clínicamente se presenta como pequeñas pápulas hiperqueratósicas distribuidas por toda la superficie palmo-plantar, pudiendo agruparse en localizaciones de más presión adoptando un patrón más difuso en esas zonas. En casos más extremos pueden adoptar un aspecto verrucoso o de cuernos cutáneos. Las alteraciones de las uñas son frecuentes, en forma de engrosamiento ungueal, discromía, hemorragias, onicogrifosis, onicomadesis, onnicorrexis, etc. Otra característica relevante de esta enfermedad es su potencial asociación con diversas neoplasias, como adenocarcinoma de colon, cáncer de próstata, cáncer de pulmón, cáncer de mama, etc. El diagnóstico diferencial incluye la exposición a arsénico, verrugas vulgares (tampoco era un diagnóstico tan descabellado al fin y al cabo), acroqueratoelastoidosis de Costa, otros tipos de queratodermia punctata, hiperqueratosis reactiva, … Si realizamos una biopsia, la histología revelará un engrosamiento de la capa córnea, aplanamiento de las estructuras subyacentes y otras alteraciones inespecíficas, como paraqueratosis y acantosis. Además de la asociación a neoplasias, también se ha descrito asociación con psoriasis, queratosis pilaris y artropatía HLA-B27.
Respecto al tratamiento, consiste en administrar agentes queratolíticos, como ácido salicílico tópico (especialmente en aquellos casos asintomáticos). Los retinoides sistémicos son una alternativa en casos más extremos que cursen con molestias. Eso fue lo que le hicimos a Fermín, después de asegurarnos con una analítica de que no existían contraindicaciones, le empezamos acitretina a dosis de 25 mg diarios, con importante mejoría a partir del primer mes. Aunque no le causó ningún problema, el tratamiento se interrumpió a los 6 meses, con recidiva de las lesiones (cosa que no sorprendió a nadie). No volvimos a ver a Fermín, pero sí supimos algunos años más tarde que había fallecido a consecuencia de un cáncer de colon.
Artículo original: http://www.dermapixel.com/2016/11/el-apasionante-mundo-de-las.html