Bei ALS wird das schädigende Protein TDP-43 von Neuron zu Neuron über Axone weitergegeben. Forscher beschreiben nun zudem typische Schädigungsmuster im zentralen Nervensystem. Ihre Studienergebnisse könnten dabei helfen, den Krankheitsverlauf besser zu verstehen.
Bei der tödlichen Nervenkrankheit Amyotrophe Lateralsklerose (ALS) kommt es zu einem fortschreitenden Untergang der bewegungskontrollierenden Nervenzellen in Gehirn und Rückenmark. Die Folge: zunehmende Lähmungen und schließlich der Tod – oft verursacht durch den Ausfall der Atemmuskulatur. Wie sich die Schädigung typischerweise im zentralen Nervensystem ausbreitet, haben Forscher aus Ulm und Philadelphia untersucht. In ihrem Fachartikel in „Annals of Neurology“ beschreiben sie, dass das Protein TDP-43 offenbar von Neuron zu Neuron weitergegeben wird. Dieses Eiweiß lagert sich bei der ALS in Nervenzellen ab. Bei ihren Untersuchungen konnten die Wissenschaftler typische Schädigungsmuster identifizieren – vom Motorcortex über weiter im Stirnhirn liegende Hirnregionen bis in den Schläfenlappen – und den Verlauf der neuropathologischen Veränderungen in Stadien einteilen. Die Erkenntnisse der Ulmer Forscher um Professor Heiko Braak, Dr. Dr. Kelly Del Tredici, Professor Johannes Brettschneider, sowie Professor Albert Ludolph helfen nicht nur, den Krankheitsverlauf besser zu verstehen. Sie könnten auch neue Ansatzpunkte für die bisher unbefriedigende medikamentöse Behandlung der ALS aufzeigen.
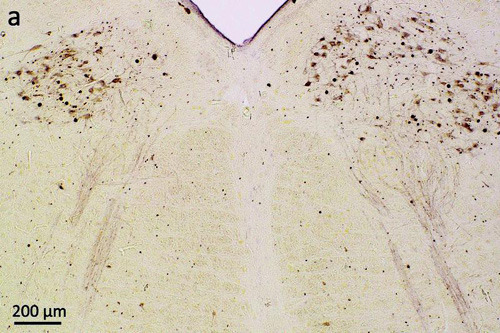
Bereits 2006 hat die Gruppe um Professorin Virginia Lee und Professor John Trojanowski, Mitautoren des jetzt erschienenen Beitrags, TDP-43 als wichtigsten Bestandteil von Ablagerungen in Nervenzellen bei ALS-Patienten beschrieben: Das Eiweiß verursacht „Verklumpungen“ in der Zelle und führt schließlich zu ihrem Untergang. Jetzt konnte die deutsch-amerikanische Forschergruppe um den Ulmer Erstautoren Johannes Brettschneider zeigen, dass sich das Protein wohl über Axone im zentralen Nervensystem von Neuron zu Neuron ausbreitet. „Der typische von uns beobachtete Verlauf der Pathologie bei ALS vom Motorcortex über den Frontallappen könnte erklären, warum ein Teil der Patienten zusätzlich Symptome einer frontotemporalen Lobärdegeneration mit Demenz entwickelt“, sagt Johannes Brettschneider. Jetzt könne man nach Wegen suchen, die Weitergabe des Proteins pharmakologisch zu unterbinden. TDP-43 Ablagerungen in den für die Bewegung der Zunge zuständigen Hirnnervenkernen des Hirnstammms © David Ewert
Für den Fachartikel haben die Forscher Proben aus 22 Hirnregionen von 76 amerikanischen ALS-Patienten immunhistochemisch gefärbt und unter dem Mikroskop untersucht. Als weiteres wichtiges Ergebnis der Studie gelang es der Gruppe, den typischen Verlauf der ALS in vier vorläufige Stadien einzuteilen. Der Anatom Heiko Braak hat weltweit verwendete Stadien entwickelt, die eine Klassifikation des Verlaufs der neurodegenerativen Krankheiten wie Alzheimer und Parkinson ermöglichen. In einem nächsten Schritt gilt es, die ALS-Stadien bei lebenden Patienten zu erkennen und ihre Behandlung entsprechend auszurichten. „Dafür werden an unserer Klinik verschiedene Ansätze beforscht. So gibt es zum Beispiel Bemühungen, typische Schädigungen im Kernspintomographen zu identifizieren“, sagt Albert Ludolph, Ärztlicher Direktor der Ulmer Universitätsklinik für Neurologie. Originalpublikation: Stages of pTDP-43 pathology in amyotrophic lateral sclerosis Johannes Brettschneider et al.; Annals of Neurology, doi: 10.1002/ana.23937, 2013