Unser Körper ist äußerlich symmetrisch. Im Körperinneren jedoch herrscht Asymmetrie. Das gilt für die meisten Menschen. Doch bei einigen von ihnen wurde diese Asymmetrie nie richtig etabliert (Situs-Anomalie) und die normale Anordnung der inneren Organe ist verändert.
Bei der Heterotaxie, einer besonderen Form der Situs-Anomalie, suchen sich die inneren Organe scheinbar unabhängig voneinander einen Platz im Bauchraum, so dass in Folge der neu entstandenen „Ordnung“ eine Art „Symmetrie“ entsteht, was auch zur Ausbildung falscher Verbindungen zwischen den Organen führen kann. Am deutlichsten wird das beim Herz. Es ist das erste Organ, das sich im Embryo entwickelt und seine Funktion erfüllen muss; damit aber auch das Organ, bei dem die meisten Entwicklungsdefekte auftreten. Patienten mit Heterotaxie leiden daher meist unter angeborenen Herzfehlern, die sehr kompliziert sein können. Wie und warum solche schweren Defekte entstehen, war bislang unklar. Dr. Martin Burkhalter vom Leibniz-Institut für Altersforschung – Fritz-Lipmann-Institut (FLI) in Jena und Dr. Melanie Philipp von der Universität Ulm gelang es nun in Zusammenarbeit mit Forschern der renommierten Duke University in den USA einen bis dato unbekannten Mechanismus zu identifizieren, der bei der Embryonalentwicklung die Asymmetrie-Ausbildung des Herzen maßgeblich beeinflusst.
„Lange bevor sich überhaupt das erste Organ in unserem Körper entwickelt, wird die Asymmetrie festgelegt“, berichtet Dr. Burkhalter. Dafür ist ein kleines Vesikel (Zellbläschen) zuständig. „Dieses vermutlich 3 Wochen nach der Befruchtung gebildete Vesikel existiert nur relativ kurz, hat aber eine ganz wichtige Funktion“, so Burkhalter weiter. „Es bestimmt über die Zilien, das sind kleine Flimmerhaare auf der Oberfläche des Vesikels, welche Seite des Körpers links und welche rechts ist und legt so fest, wo später das Herz liegt und wie es über Blutgefäße mit der Lunge und dem Rest des Körpers verbunden ist.“
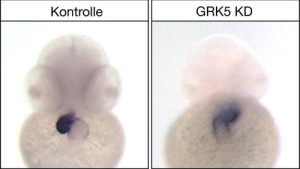
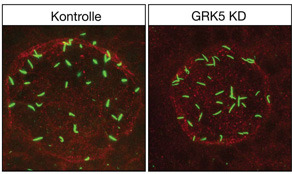
Die Wissenschaftler konnten nun am Modellorganismus Zebrafisch nachweisen, dass GRK5 (G-Protein-gekoppelte Rezeptor-Kinase 5) entscheidenden Einfluss auf die Ausbildung der Asymmetrie des Herzen während der embryonalen Entwicklung hat. „Bereits früher war der Einfluss von GRK5 auf die Funktion des adulten, ausgewachsenen Herzen diskutiert und nachgewiesen worden“, so Dr. Melanie Philipp, „doch bislang wurde ihr Einfluss bei der Embryonalentwicklung noch nicht detailliert untersucht.“ Unten links: Das Herz des Zebrafischemrbyos entwickelt sich auf der linken Seite. Unten rechts: Im GRK5-Knockdown verläuft die Entwicklung spiegelverkehrt. Die Färbung wurde jeweils durch eine so genannte in situ Hybridisierung realisiert. © Uni Ulm „Uns gelang es, einen neuartigen Mechanismus zu identifizieren, wie GRK5 die Ausbildung der Asymmetrie reguliert“, erläutert Dr. Burkhalter. GRK5 wirkt dabei direkt in dem temporär auftretenden Asymmetrie-Vesikel und hält dort den mTOR-Signalweg, ein zentraler Regulator des Zellwachstums, in einer Art Gleichgewicht (Homöostase). Wird dieses Gleichgewicht zum Beispiel durch verhinderte Expression von GRK5 gestört, nimmt die mTOR-Signalaktivität zu. Mit fatalen Folgen: Die Zilien auf dem Vesikel verändern sich und werden länger. Abgebildet sind die Kupfferschen Vesikel, also das Lateralitätsorgan. In der grünen Färbung wird sichtbar, dass die Zilien im Knockdown länger sind (Aufnahme in gleicher Vergrößerung). Deren Bewegungen bestimmen indirekt darüber, wo im Organismus später rechts und links angelegt wird. Konfokalmikroskopie: © M. Philipp „Eine geänderte Gestalt der Zilien verändert in der Regel auch den Fluss, der durch die Flimmerbewegungen der Zilien entsteht, was wiederum zu falschen Signalen für die Ausbildung einer Asymmetrie und damit für die normale Entwicklung des Herzens führt.“ Die Ergebnisse dieser Grundlagenforschung besitzen auch medizinisches Potential: GRK5 könnte als Suszeptibilitäts-Allel für bestimmte Fälle von angeborenen Herzkrankheiten fungieren. Originalpublikation: Grk5l Controls Heart Development by Limiting mTOR Signaling during Symmetry Breaking Martin D. Burkhalter et al.; Cell Reports, doi:10.1016/j.celrep.2013.07.036 ; 2013