Um Heterogenitäten von Zellen zu verstehen, sind Einzelzell-Analysen nötig. Diese sind jedoch aufwändig und oft ungenau. Wissenschaftler haben nun einen Weg gefunden, wie sich die Analysen durch mathematische Methoden vereinfachen lassen.
Jede unserer Körperzellen ist einzigartig. Selbst Zellen einer Gewebeart, die unter dem Mikroskop völlig gleich aussehen, unterscheiden sich geringfügig voneinander. Um zu verstehen, wie sich aus einer Stammzelle eine Herzzelle entwickeln kann, warum die eine Beta-Zelle Insulin produziert und die andere nicht und warum eine normale Gewebezelle plötzlich zu einer Krebszelle wird, untersuchen Wissenschaftler seit einigen Jahren gezielt die Aktivitäten der RNA. Permanent werden Stoffe auf- und abgebaut. Ständig lesen RNA-Moleküle Baupläne für Eiweiße aus der Erbsubstanz ab und lassen sie von anderen Eiweißen produzieren. Inzwischen hat die Wissenschaft die Methoden so verfeinert, dass es möglich ist, alle in einer einzigen Zelle zu einem bestimmten Zeitpunkt aktiven RNA-Moleküle zu erfassen. Ende Dezember 2013 wurde die Einzelzell-Sequenzierung vom Fachblatt Nature Methods zur Methode des Jahres 2013 erklärt. Doch die Untersuchung einzelner Zellen ist extrem aufwändig und die Handhabung der Zellen verursacht Fehler und Ungenauigkeiten, die ein erhebliches statistisches Rauschen zur Folge haben. Vor allem schwächere Unterschiede in der Genregulierung gehen darin unter oder werden gar nicht erst sichtbar.
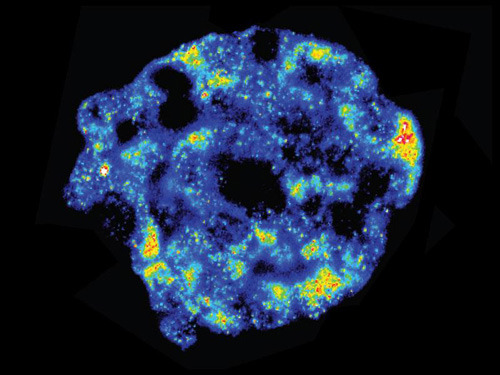
Wissenschaftler unter der Leitung von Professor Fabian Theis, Inhaber des Lehrstuhls für Mathematische Modelle biologischer Systeme der TU München und Leiter des Instituts für Computational Biology am Helmholtz Zentrum München, haben nun einen Weg gefunden, wie sie mit Methoden der mathematischen Statistik die Einzelzell-Analyse wesentlich verbessern können. Statt nur jeweils einer Zelle nahmen sie zwischen 16 und 80 Proben mit jeweils zehn Zellen. „Eine Menge von zehn Zellen ist wesentlich leichter zu handhaben“, sagt Professor Theis. „Bei der zehnfachen Menge an Zellmaterial werden die Umgebungseinflüsse deutlich zurück gedrängt.“ Allerdings sind die Zellen mit unterschiedlichen Eigenschaften dann zufällig über die Proben verteilt. Daher entwickelte Theis´ Mitarbeiterin Christiane Fuchs statistische Methoden, um die Einzelzell-Eigenschaften dennoch zu identifizieren. Fluoreszenz-in-situ-Hybridisierung zeigt mRNA-Aktivität. Blau: niedrige, rot: hohe Aktivität.© S. S. Bajikar / University of Virginia, Charlottesville (USA)
Auf der Basis bekannter biologischer Daten modellierten Theis und Fuchs die Verteilung für den Fall von Genen, die sich in zwei definierten regulatorischen Zuständen befinden können. Zusammen mit den Biologen Kevin Janes und Sameer Bajikar von der Universität Virginia in Charlottesville (USA) konnten sie experimentell belegen, dass mit Hilfe der statistischen Methoden aus den Messergebnissen der zehn Zellen enthaltenden Proben genauere Ergebnisse errechnet werden können als mit der Analyse der gleichen Anzahl von Einzelzellproben. In vielen Fällen werden durch einen Faktor gleich mehrere Genreaktionen angestoßen. Auch auf solche Fälle ließ sich das statistische Verfahren anwenden. Fluoreszierende Marker zeigen die Genaktivitäten, und man erhält so ein Mosaik aus dem sich wiederum ablesen lässt, ob verschiedene Zellen unterschiedlich auf den Faktor reagieren. Die Methode ist so empfindlich, dass sie selbst eine Abweichung unter 40 sonst gleichen Zellen noch zeigt. Dass diese Abweichung tatsächlich ein Effekt ist und nicht ein zufälliger Ausreißer, konnte experimentell belegt werden. Originalpublikation: Parameterizing cell-to-cell regulatory heterogeneities via stochastic transcriptional profiles Fabian Theis et al.; PNAS, doi: 10.1073/pnas.1311647111, 2014