Einem Forscherteam ist es nun gelungen, eine gefährliche und bislang kaum bekannte Stoffwechselkrankheit zu entschlüsseln. Zudem konnten sie eine Therapiemöglichkeit gleich mit etablieren und in die klinische Anwendung bringen.
Es passierte in einer süddeutschen Stadt: Ein Schüler hastet dem Bus nach, in Sorge, ihn zu verpassen. Nach Atem ringend, erreicht er den Bus knapp, bricht darin aber plötzlich tot zusammen. Der junge Mann kann wiederbelebt werden, behält aber bleibende Schäden zurück. Die diesem realen Fall zugrunde liegende, bislang kaum bekannte Stoffwechselkrankheit hat nun ein deutsch-niederländisches Forscherteam unter Leitung von Wissenschaftlern der Universität Münster entschlüsselt. Eine Therapiemöglichkeit konnten die Forscher gleich mit etablieren und in die klinische Anwendung bringen.
Den Betroffenen fehlt das Enzym Phosphoglucomutase 1 (PGM 1), das im Zellplasma jeder Zelle vorkommt und für die Speicherung von Energie aus Glukose, also Zucker, im Essen zuständig ist. Außerdem setzt es Energie frei, wenn der Körper – zum Beispiel bei plötzlicher Anstrengung – auf seine Glykogenreserven zurückgreifen muss. „Dass den Patienten dieses Enzym fehlt, kann viele schwerwiegende Folgen haben“, erläutert Prof. Thorsten Marquardt, der in der Klinik für Kinder- und Jugendmedizin des Universitätsklinikums Münster den Bereich für angeborene Stoffwechselerkrankungen leitet. „Bei PGM1-Mangel kann es beispielsweise zu Muskelschmerzen und Muskelzerfall, rotem Urin nach dem Sport, Lebererkrankungen, einem gefährlich niedrigen Blutzuckerwert und sogar schweren Herzmuskelerkrankungen kommen.“ So unterschiedlich diese Symptome sind, gründen sie doch auf einer Ursache: dem PGM1-Mangel. Diesen haben Marquardt und seine Kollegen gemeinsam mit einem Team um Dr. Dirk Lefeber von der Universitätsklinik Nijmegen bei 19 Patienten untersucht.
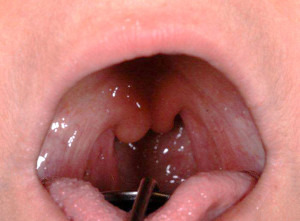
Obwohl die Krankheit so viele „Gesichter“ hat, damit schwer zu diagnostizieren und besonders gefährlich ist, gibt es eine recht einfache Diagnosemöglichkeit, wie Marquardt berichtet: „Hinten am Gaumen hat jeder Mensch ein Zäpfchen. Bei Menschen mit PGM1-Mangel ist dieses gespalten und sieht dann wie zwei Zäpfchen aus.“ Ein simpler Blick in den Spiegel könne also Hinweise auf eine schwerwiegende Erkrankung mit potentiell tödlichen Folgen liefern, so der Marquardt. Er und seine Kollegen hoffen deshalb, dass die Untersuchung des Zäpfchens Eingang in die Vorsorgeuntersuchungen bei Kindern findet. „Mit einem speziellen, von uns entwickelten Biomarker ließe sich diese Diagnose dann anhand eines einfachen Bluttests bestätigen“, so Dirk Lefeber. Das Risiko, an den Folgen der Erkrankung, beispielsweise am plötzlichen Herztod, zu sterben, würde so erheblich minimiert – denn auch die Behandlung ist erstaunlich unkompliziert. Als Uvula bifida – hier bei einer Patientin – bezeichnen Mediziner die Längsspaltung des Gaumenzäpfchens. Sie kann ein Hinweis auf einen PGM1-Mangel sein. © S. Anghelescu
Wie erfolgreiche klinische Tests der Forschergruppe nämlich zeigen, lässt sich der Enzymmangel durch Gabe von Galaktose nahezu ausgleichen. Die Hauptquelle von Galaktose ist Laktose, besser bekannt als Milchzucker, den es in jedem Supermarkt zu kaufen gibt. Das Therapiekonzept der Wissenschaftler umfasst zudem, den Ernährungsplan um spezielle Kohlenhydrate zu ergänzen und auf besonders energetisches Sporttreiben zu verzichten – auch dies Maßnahmen, die keinen besonderen Aufwand erfordern. Die Erkenntnisse der Forscher werden bereits in der klinischen Praxis eingesetzt: Am Universitätsklinikum Münster profitieren davon erste Patienten in der Klinik für Kinder- und Jugendmedizin. Für die Zukunft erhoffen sich Marquardt, Lefeber und ihre Kollegen aus Münster und Nijmegen, dass sich das Wissen um diese gefährliche Krankheit, die jedoch so einfach zu diagnostizieren und zu behandeln ist, bei den Berufskollegen rasch verbreitet. Originalpublikation: Multiple Phenotypes in Phosphoglucomutase 1 Deficiency Laura C. Tegtmeyer et al.; New England Journal of Medicine, doi: 10.1056/NEJMoa1206605; 2014