Wissenschaftler fanden nun heraus, dass eine schwere Form der Epilepsie bei Kleinkindern durch bislang unbekannte Mutationen am Ionenkanal HCN1 ausgelöst wird. Die Veränderungen am Erbgut entstehen neu, sind bei den Eltern also nicht nachweisbar.
Epileptische Enzephalopathien sind schwere Krankheiten, die schon bei Babys auftreten. Sie gehen mit einer gestörten Reifung des Gehirns sowie mit einer Beeinträchtigung der geistigen und manchmal auch der motorischen Entwicklung einher. Meistens treten die Krampfanfälle zuerst in Zusammenhang mit Fieber auf und lassen sich in der Regel nicht therapieren. Eine schon länger bekannte Form der frühkindlichen epileptischen Enzephalopathie ist das Dravet-Syndrom. Es tritt bei etwa einem von 30.000 Kindern auf und wird durch Mutationen am Natriumkanal-Gen SCN1A ausgelöst. Bei vielen Kindern, deren Krankheit dem Dravet-Syndrom ähnelt, liegen jedoch keine Mutationen in SCNA1 vor. Es müssen also andere Gene für diese frühkindliche Form der Epilepsie verantwortlich sein.
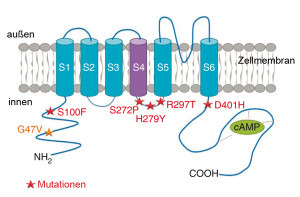
Auf der Suche nach neuen Krankheitsgenen für frühkindliche epileptische Enzephalopathien sind Wissenschaftler aus Paris und Würzburg nun fündig geworden. Im Erbgut von fast 200 betroffenen Kindern, bei denen Mutationen im SCNA1-Gen bereits ausgeschlossen waren, entdeckten sie in sechs Fällen die krankheitsverursachenden Mutationen an einem anderen Ionenkanal-Gen, bei HCN1. Diese dominanten Mutationen entstehen bei der Bildung der elterlichen Keimzellen jeweils neu; in den Körperzellen der Eltern sind sie nicht vorhanden. „Der von dem Kationenkanal HCN1 getragene elektrische Strom wird auch als ‚Schrittmacher‘ bezeichnet, da er in spontan aktiven Nervenzellen die rhythmische Aktivität fördert“, sagt Professor Thomas Haaf, Leiter des Instituts für Humangenetik der Universität Würzburg. Tiermodelle hätten bereits vermuten lassen, dass dieser Kanal bei Epilepsien eine Schlüsselrolle spielt. „Aber bei Patienten waren bisher keine entsprechende Mutationen gefunden worden.“ Struktur des Kanals HCN1 in der Zellmembran von Nervenzellen. Die Sterne kennzeichnen Stellen, an denen Mutationen entdeckt wurden. Die meisten davon aktivieren den Kanal wahrscheinlich. © Institut für Humangenetik
Die Krampfanfälle bei Kindern mit Mutationen im HCN1-Gen sind anfangs kaum vom Dravet-Syndrom zu unterscheiden, im weiteren Verlauf aber schon: „Es kommt dann vermehrt zu atypischen Anfällen. Alle betroffenen Kinder zeigen eine Intelligenzminderung und Verhaltensstörungen, autistisches Verhalten eingeschlossen“, so Professor Haaf.
Wie lässt sich das neue Wissen zum Wohl der Patienten nutzen? Diese Frage ist nicht einfach zu beantworten, denn von der Entdeckung einer krankheitsverursachenden Genmutation bis zur Therapie ist es meistens ein langer Weg. Professor Haaf dazu: „Auf jeden Fall hilft ein besseres Verständnis der molekularen Krankheitsursachen bei der Entwicklung neuer therapeutischer Ansätze, zum Beispiel von Medikamenten, welche die HCN1-Ströme spezifisch beeinflussen. Schon jetzt ermöglichen die Ergebnisse, bei dieser meist sporadisch auftretenden sehr schweren frühkindlichen Erkrankung die richtige Diagnose zu stellen und die Eltern bei der weiteren Familienplanung genetisch zu beraten.“ Originalpublikation: De novo mutations in HCN1 cause early infantile epileptic encephalopathy Caroline Nava et al.; Nature Genetics, doi: 10.1038/ng.2952; 2014