Die tief hängenden Früchte der Medikamentenentwicklung sind geerntet. Um an bisher unerreichbare Targets heranzukommen, müssen neue Wirkprinzipien her. Können molekulare Klebstoffe die Lücke schließen?
Derzeit gibt es rund 4.500, insbesondere von der FDA zugelassene Medikamente. Davon handelt es bei ca. 2.800 um sogenannte Small Molecules – also klassische, chemisch synthetisierte niedermolekulare Substanzen – und bei knappen 1.700 um Biologika (beispielsweise Antikörper oder Peptide). Und jedes Jahr kommen ca. 50 bis 60 Neuzulassungen hinzu. All diese Pharmaka, inklusive derjenigen in der Entwicklung, adressieren als Agonisten, Antagonisten oder Modulatoren knappe 5.300 nicht-redundante Proteintargets. Dazu gehören unter anderem humane Rezeptoren, Enzyme, Ionenkanäle oder Transporter sowie virale bzw. bakterielle Zielstrukturen im Falle von Antibiotika und Virostatika.
Das humane Genom mit seinen drei Milliarden Basenpaaren enthält ca. 20.000 proteinkodierende Gene und damit – ohne Berücksichtigung von Isoformen – den Bauplan für die gleiche Anzahl an Proteinen. Durch die aktuell präklinisch sowie klinisch vorhandenen niedermolekularen Wirkstoffe und Biologika können jedoch nur knappe 10 % des humanen Proteoms spezifisch bzw. selektiv pharmakologisch beeinflusst werden. Der weitaus größere Anteil ist als sogenanntes Dark Proteom derzeit nicht direkt für Medikamente zugänglich. Diese Grenze ist unter anderem dadurch bedingt, dass viele Targets, also pharmakologische Zielstrukturen, als „undruggable“ gelten. Sie können im Rahmen einer Medikamentenentwicklung technisch nicht adressiert werden, da ca. 75 % der zellulären Proteine in ihrer dreidimensionalen Struktur keine Taschen aufweisen, an welche Medikamentenmoleküle mit ausreichender Affinität binden könnten. Insbesondere Transkriptionsfaktoren gelten als kaum druggable.
In diesem Kontext ist z. T. auch vom low-hanging-fruit-Problem die Rede, welches impliziert, dass es sich bei den bisher verfügbaren Medikamenten und ihren Targets um die einfach zu pflückenden Früchte gehandelt hat. Um einen größeren Anteil unseres Genoms bzw. Proteoms für Pharmaka zu erschließen, sind daher neue Methoden in der Medikamentenentwicklung notwendig.
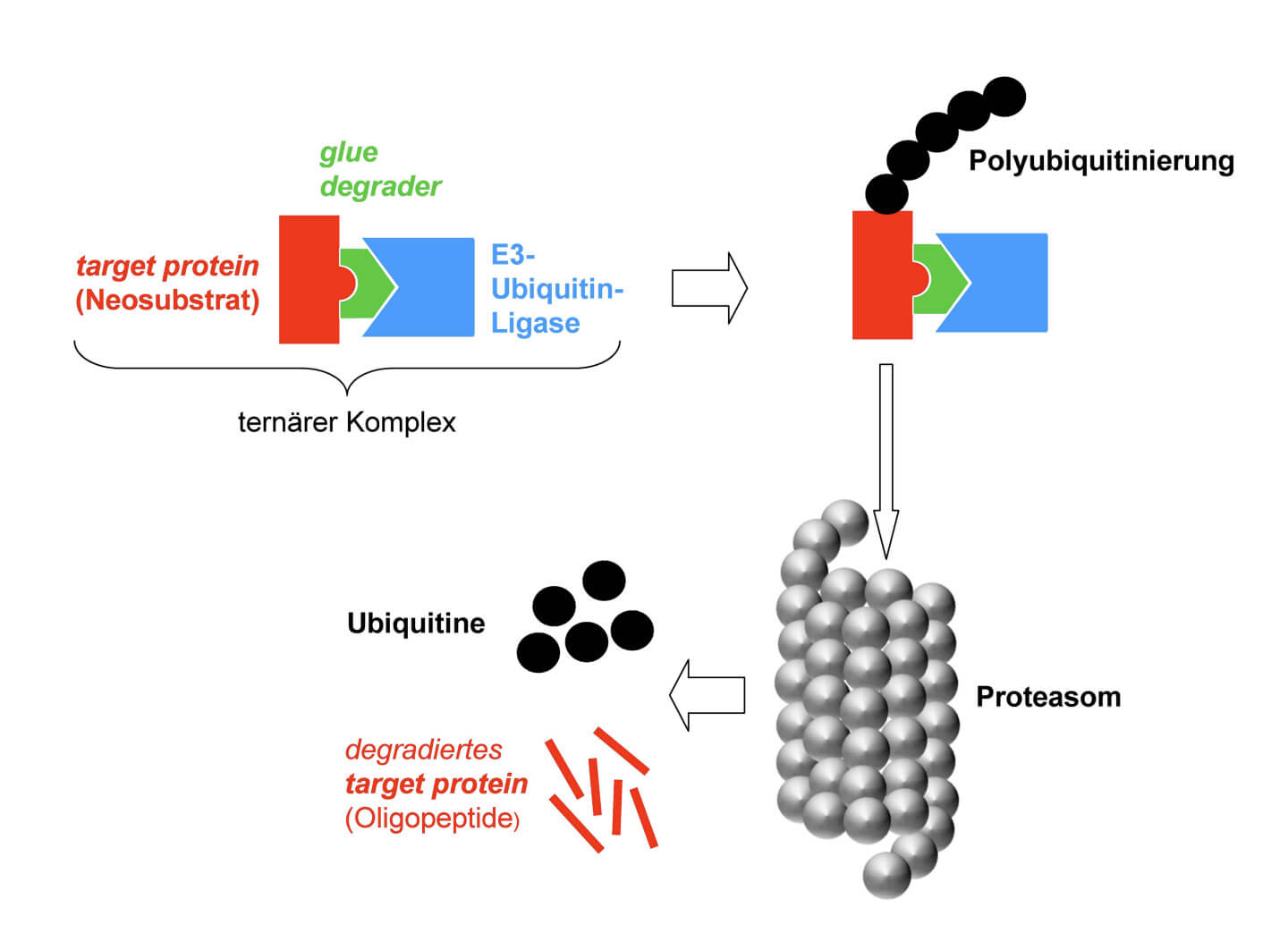
Und nun kommen die sogenannten Molecular Glue Degrader (deutsch: molekulare Klebstoff-Degradierer) ins Spiel, welche das Potenzial haben, ansonsten als undruggable betrachtete Proteine spezifisch zu degradieren. Es handelt sich dabei um Medikamentenmoleküle, die Proteine mit einer sogenannten Ubiquitinligase verkleben. Das führt im Endeffekt dazu, dass dieses Protein im Proteasom – der Müllabfuhr der Zelle – abgebaut bzw. degradiert wird.Der Wirkmechanismus von Glue Degraders. Credit: Funke-Kaiser
Die Geschichte der Molecular Glue Degrader ist eng verknüpft mit Thalidomid (Contergan®). Dieses Pharmakon war primär ein Schlafmittel, wurde jedoch auch als Antiemetikum in der Schwangerschaft eingesetzt. Nach nur vierjährigem Vertrieb wurde es in Deutschland 1961 aufgrund der bekannten, schwerwiegenden teratogenen Effekte (Phokomelie-Dysmelie-Syndrom) wieder vom Markt genommen. In den USA war Thalidomid interessanterweise nie als Schlafmittel zugelassen.
In den 90er-Jahren wurde die Wirkung von Thalidomid bei Lepra sowie seine Antitumorwirkung beim multiplen Myelom erkannt, woraufhin es 1998 bzw. 2006 für diese Indikationen zugelassen wurde. Es besitzt eine immunmodulatorische und antiangiogenetische Aktivität. Lenalidomid (Revlimid®), ein Nachfolgepräparat von Thalidomid mit hoher chemischer Strukturähnlichkeit, ist in Deutschland beim multiplen Myelom, beim Mantelzelllymphom und bei myelodysplastischen Syndromen jeweils als Monotherapie zugelassen. Der molekulare Wirkmechanismus beider Substanzen war jedoch bis vor Kurzem unbekannt.
2010 konnte eine japanische Arbeitsgruppe zeigen, dass Thalidomid eine bestimmte E3-Ubiquitinligase, genannt Cereblon, bindet. Weiterhin konnte demonstriert werden, dass Lenalidomid, dessen molekulares Target auch Cereblon ist, eine Ubiquitinierung und anschließende Degradation der beiden Transkriptionsfaktoren Aiolos und Ikaros verursacht. Dies ist wiederum mit einer konsekutiven Induktion von Interleukin-2 und einer T-Zell-Aktivierung verbunden. Der erste, 2007 beschriebene Glue Degrader war jedoch das Pflanzenhormon Auxin.
Die neue Medikamentenklasse der Glue Degraders ist dadurch charakterisiert, dass eine bereits physiologisch vorhandene, aber schwache Protein-Protein-Interaktion verstärkt wird. Es bildet sich ein sogenannter ternärer Komplex ("Dreierkomplex") aus dem niedermolekularen Medikament (z. B. Lenalidomid), einer E3-Ligase und einem Zielprotein (auch Neosubstrat genannt), das spezifisch abgebaut werden soll. Dadurch kommt es zur Polyubiquitinierung des Zielproteins und seiner anschließenden Degradation im Proteasom.
Bildlich kann man sich dies als Puzzle vorstellen: Ein Puzzlestück wird durch den Kleber an seinen Kanten so modifiziert, dass es nun perfekt in ein anderes Puzzleteil hineinpasst, zu welchem es zuvor nicht bzw. kaum komplementär war.
Die Komplexität dieses biochemischen Systems, aber auch das Potenzial hinsichtlich der Medikamentenentwicklung wird auch dadurch deutlich, dass unser Genom für mehr als 600 E3-Ligasen kodiert. Daher haben zahlreiche Pharma- und Biotechfirmen (u. a. Novartis, Roche, Amgen und Monte Rosa Therapeutics) Molecular Glue Degraders in ihren Pipelines.
Beispielsweise befindet sich die Substanz MRT-2359 derzeit in der Phase I/II der klinischen Prüfung u. a. für die Indikationen Bronchialkarzinom (NSCLC und SCLC), Mammakarzinom und Prostatakarzinom. Der Glue Degrader PLX-4545, welcher die E3-Ligase Cereblon rekrutiert und den vermeintlich undruggable Transkriptionsfaktor IKZF2 angreift, befindet sich in Phase I. Die geplanten Indikationen sind solide Tumore. Auch in Phase I befindet sich ein Degradierer des Translations-Terminationsfaktors GSPT1, der bei Myc-abhängigen Tumoren (z. B. Kolon- und Mammakarzinom) zum Einsatz kommen soll.
Weitere onkologisch relevante Targets für Glue Degraders, die derzeit verfolgt werden, sind die Cyclin-abhängige Kinase (CDK) und der Splicing-Faktor CAPERalpha (RBM39). Aber auch nicht-onkologische Erkrankungen wie z. B. die Sichelzellanämie werden derzeit hinsichtlich der klinischen Anwendung von Glue Degraders verfolgt.
Die Gesamtbedeutung dieser neuen Medikamentenklasse ist auch daran ersichtlich, dass die Deal-Volumina durch Lizenzvereinbarungen und Firmenübernahmen aktuell sogar die entsprechenden finanziellen Vereinbarungen von Antikörpern, anderen Small Molecules und Gentherapeutika überstiegen haben. Aufgrund dieser Investitionen und des technisch-innovativen Potenzials ist es insgesamt sehr wahrscheinlich, dass in den nächsten Jahren neue Glue Degraders zugelassen werden – und zwar für Indikationen, die mit den bisherigen pharmakologischen Ansätzen nicht oder kaum adressierbar waren.
Bildquelle: Anyzoy studio, Unsplash