Bislang werden bei Mukoviszidose vor allem Symptome behandelt. Jetzt gibt es Ergebnisse zu einer Kausaltherapie: Tezacaftor, ein neuer „CFTR-Korrektor“, kann in Kombination mit Ivacaftor die Lungenfunktion von Patienten verbessern. Wirklich ein Fortschritt?
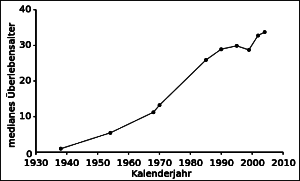
In Deutschland leidet eins von 2.500 Neugeborenen an Mukoviszidose. Mittlerweile sind Screenings auf diese am häufigsten tödlich verlaufende angeborene Stoffwechselkrankheit zur Regelleistung geworden. Ärzte suchen u.a. nach Mutationen im CFTR-Gen (Cystic Fibrosis Transmembrane Conductance Regulator). Bei Erkrankten bilden manche Zellen nur noch defekte Chloridkanäle aus. Gelangt zu wenig Wasser aus dem umgebenden Gewebe in exokrine Drüsen, werden die Sekrete zähflüssig und Organe erfüllen ihre Aufgaben nicht mehr richtig. In den letzten Jahrzehnten ist es gelungen, Patienten mit Pharmaka, Sport, Physiotherapien und Diäten immer besser zu versorgen. Der Erfolg ist nicht von der Hand zu weisen. Trotzdem liegt die Lebenserwartung kaum bei über 40 Jahren
Das liegt an der Beeinträchtigung unterschiedlicher Organsysteme. Besonders häufig treten Probleme in den Bronchien auf. Das Flimmerepithel kann den zähen Schleim nicht richtig abtransportieren. Patienten leiden an Husten und an wiederkehrenden, schweren Infektionen. Im Biofilm fühlen sich Bakterien wie Pseudomonas aeruginosa, Staphylococcus aureus, Burkholderia cepacia und Haemophilus influenzae heimisch. Es kommt zu rezidivierenden Bronchitiden und Pneumonien. Typisch sind auch Bronchiektasen, also starke Ausweitungen der Bronchien und entzündlichen Prozessen. Zunahme des medianen Überlebens bei Mukoviszidose © Kuebi / Wikipedia, CC0 Zur Schleimlösung sollten Patienten regelmäßig Kochsalzlösung, Acetylcystein oder Ambroxol inhalieren. Als Zusatz hat sich das Enzym rekombinante humane DNase bewährt, um Bestandteile neutrophiler Granulozyten im Schleim zu spalten. Sie gehen auf bereits abgeklungene Infektionen zurück. Werden die Fragmente abgebaut, verringert sich auch die Viskosität. Gegen rezidivierende Infektionen verordnen Ärzte hochdosierte Antibiotika. Bronchodilatatoren, etwa Ipratropiumbromid, Salbutamol oder Fenoterol, verbessern die Lungenfunktion. Bei zunehmender Lungeninsuffizienz bleibt nur noch die Sauerstoff-Langzeittherapie. Auch die Bauchspeicheldrüse arbeitet bei Mukoviszidose nicht richtig. Patienten leiden an chronischen Durchfällen und Fettstühlen. Da Nährstoffe nicht richtig verwertet werden, steht Untergewicht auf der Tagesordnung. Hochkalorische Kost verbessert in Kombination mit Verdauungsenzymen die Situation. Kommt es zum Ileus, ist chirurgische Hilfe erforderlich. Da sich zunehmend Bindegewebe in den Pankreas einlagert, kann es zum CF-assoziierten Diabetes (cystic fibrosis related diabetes, CFRD) kommen, einer Form des Typ 3-Diabetes. Endokrinologen verordnen möglichst rasch Insulin. Warum bei Mukoviszidose Osteoporose als Spätkomplikation auftritt, lässt sich nicht abschließend beantworten. Es liegt nicht nur an der Malabsorption. Knochenabbauende Osteoklasten sind deutlich aktiver als bei gesunden Menschen. Ihre Gegenspieler, die knochenaufbauenden Osteoblasten, stellen defektes CFTR-Protein her. Das führt vermutlich zu Störungen des Gleichgewichts zwischen Osteoprotegerin und Prostaglandin E2 und folglich zur stärkeren Knochenresorption. Therapeutisch eignen sich Wirkstoffe aus der Osteoporose-Therapie, beispielsweise Bisphosphonate, Strontiumranelat oder Teriparatid.
Alle Ansätze verfolgen das Ziel, Symptome zu lindern. Umso größer ist der Wunsch nach kausalen Therapien. Im Jahr 2015 zeigte Eric Alton vom Royal Brompton and Harefield NHS Foundation Trust, London, dass Gentherapien prinzipiell möglich sind. Seine Ergebnisse einer Phase 2-Studie blieben jedoch weit hinter den Erwartungen zurück. Patienten berichteten im Vergleich zu Placebo nur von einer geringfügigen Verbesserung ihrer Lungenfunktion. Alton arbeitete klassisch mit einem Plasmid, um intakte CFTR-Gene in Zellen einzubringen. Ob die vieldiskutierte „Genschere“ CRISPR/Cas9 zu besseren Resultaten führt, ist offen. Derzeit bleiben Lungentransplantationen als Ausweg, wobei der langfristige Erfolg bei Wissenschaftlern umstritten ist. Aufgrund fehlender Spenderorgane eignet sich diese Behandlung ohnehin nur für schwerste Fälle.
Forschern ist es mittlerweile gelungen, Arzneistoffe zu entwickeln, um defekte CFTR-Proteine im gewissen Rahmen zu reparieren. Hier handelt es sich um patientenindividuelle Ansätze, die sich stark an der jeweiligen Mutation orientieren:
Ivacaftor © Ed (Edgar181) / Wikipedia, CC0 Im Jahr 2012 begann mit Ivacaftor die Ära der personalisierten Therapien. Das Molekül wurde bei einer seltenen Klasse 3-Mutation (G551D, also einer Mutation an Stelle 551) zugelassen. Sie ist bei etwa 0,9 Prozent aller Patienten in Deutschland nachweisbar. Ivacaftor beeinflusst den defekten Chloridionenkanal, indem es dessen längere Öffnung bewirkt. Dies hat eine erhöhte Wahrscheinlichkeit des Chloridtransports durch den Kanal zur Folge. Mit 71,8 Prozent ist die schwerwiegendere Klasse 2-Mutation Mutation ΔF508 wesentlich relevanter. An der Stelle 508 fehlt die Aminosäure Phenylalanin. Ivacaftor allein führt nicht zum gewünschten Effekt. Deshalb hatten Forscher eine Idee. Sie kombinierten zwei Arzneistoffe mit unterschiedlichen Eigenschaften. Als „Korrektor“ sorgt Lumacaftor für die passende Raumstruktur und stabilisiert das Protein. Es gelangt bis an die Zellmembran, ohne dass ein durchlässiger Kanal entsteht. Erst der „Aktivator“ Ivacaftor öffnet den Kanal. „Die Therapie hat aber mehrere Nachteile“, schreibt Hartmut Grasemann von der University of Toronto. „Der Effekt ist bescheiden, was wahrscheinlich auf Interaktionen beider Arzneistoffe zurückzuführen ist.“ Ivacaftor und Lumacaftor werden beide über Cytochrom P450-3A4-Enzme in der Leber abgebaut. Hinzu kämen Dyspnoen, Leberschäden oder Wechselwirkungen mit weiteren Arzneistoffen.
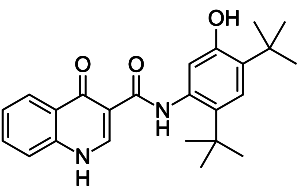
Jetzt stellen Forscher Ergebnisse zu Tezacaftor, einem neuen „Korrektor“, vor. Das Molekül wurde zusammen mit Ivacaftor in zwei herstellerfinanzierten Phase 3-Studien untersucht. Alle Patienten hatten die häufige ΔF508-Mutation. An der Studie EVOLVE nahmen 509 Patienten ab 12 Jahren teil. Sie erhielten Tezacaftor plus Ivacaftor oder Placebo. Nach 24 Wochen schlossen 475 die Studie ab. Unter Verum verbesserte sich die Lungenfunktion, gemessen als Einsekundenkapazität (FEV1), absolut um 4,0% und relativ um 6,8%. Exazerbationen traten um 35% seltener auf. Hinzu kommen Ergebnisse aus der zweiten Studie EXPAND. 162 Patienten im Alter von mindestens 12 Jahren erhielten Tezacaftor plus Ivacaftor, 157 nur Ivacaftor und weitere 162 Placebo. Nach acht Wochen verbesserte sich der FEV1-Wert gemessen an Placebo um 6,8 Prozent (Kombination), 4,7 Prozent (Ivacaftor). In beiden Studien waren unerwünschte Reaktionen meist schwach oder moderat ausgeprägt. Dazu zählten Exazerbationen mit Hämoptysen, Fieber, Kopfschmerzen, Infektionen, Entzündung der Nase und des Rachens, Fatigue sowie mehr Sputum. Allerdings bleibt sowohl bei EVOLVE als auch bei EXPAND ungeklärt, ob Tezacaftor alleine oder in Kombination mit Ivacaftor besser als die bereits zugelassene Kombination Lumacaftor/Ivacaftor wirkt. Eine Vergleichsgruppe fehlt bislang. Trotz offener Fragen hat Vertex jetzt die Zulassung in den USA und in Europa beantragt. Gleichzeitig will der Hersteller Kombinationen aus drei aktiven Molekülen untersuchen.
Die Arzeistoffe aus der Klasse der CFTR-Potentiatoren bieten nicht nur Anlass zur Euphorie. Laut Gemeinsamem Bundesausschuss sei der Zusatznutzen für Lumacaftor/Ivacaftor zwar „beträchtlich“. Dem steht allerdings der Preis in Höhe von rund 200.000 Euro für die Ein-Jahres-Therapie gegenüber. Zum Vergleich: Medikamente für eine Standardbehandlung schlagen mit 21.000 Euro pro Patient und Jahr zu Buche. Und eine Lungentransplantation kostet, Spenderorgane vorausgesetzt, etwa 150.000 Euro.