Bei einer idiopathischen Lungenfibrose stehen die Chancen schlecht: Derzeit verfügbare Therapien verlangsamen die Krankheit nur. Erfahrt hier, wie ihr Patienten trotzdem helfen könnt.
Die idiopathische Lungenfibrose (IPF) ist eine seltene, fortschreitende Lungenerkrankung. Bei Patienten vernarbt und verdickt sich das Lungeninterstitium, das Bindegewebe zwischen den Alveolen. Die genauen Pathomechanismen dieser chronisch-fibrotischen Vorgänge sind nicht bekannt. Forscher sehen die chronisch-entzündlichen Vorgänge mittlerweile eher als Folge, denn als Ursache fibrotischer Prozesse.
Zu den Details: In Europa liegt die Inzidenz der IPF bei 3–9 Fällen pro 100.000 Einwohner pro Jahr, Tendenz steigend. Die Prävalenz bewegt sich in einer Größenordnung von 30 bis 60 Fällen pro 100.000 Einwohner. Damit ist die IPF eine seltene Erkrankung, aber die häufigste interstitielle Lungenerkrankung.
„Idiopathisch“ bedeutet, dass genaue Ursachen der Erkrankung unbekannt ist. Forscher nehmen an, dass bei IPF eine Kombination von genetischen Faktoren und Umweltexpositionen eine Rolle spielen, bekannt als Gen-Umwelt-Interaktion. Zu den Umweltfaktoren gehören neben Tabak auch Noxen wie Metall- und Holzstäube oder Asbest. Weitere Risikofaktoren sind virale Infektionen, etwa mit Hepatitis oder EBV. In Familien tritt die IPF teilweise gehäuft auf, vermutlich in Form einer autosomal-dominanten Vererbung.
Typische IPF-Patienten sind deutlich älter als 50, oft Männer, meist aktive oder frühere Raucher. Zum Arzt gehen sie wegen Atemnot unter Belastung oder trockenem, chronischem Husten. Bei fortgeschrittenem Krankheitsverlauf wird die Luft schon in Ruhe knapp, Erschöpfung kommt hinzu. Außerdem leiden viele Patienten an rezidivierenden Atemwegsinfekten. Durch den hohen Energieverbrauch kommt es auch zu ungewolltem Gewichtsverlust.
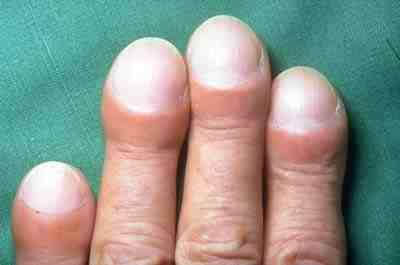
Bei Patienten mit fortgeschrittener IPF können sich die Finger und Zehen hin zu sogenannten Trommelschlegelfingern beziehungsweise Uhrglasnägeln verändern. Als Ursache gilt chronischer Sauerstoffmangel in Geweben.
Trommelschlegelfinger bei IPF. Credit: Wikimedia Commons.
Manche dieser Symptome sind recht unspezifisch. Sie deuten ebenfalls auf eine chronisch obstruktive Lungenerkrankung (COPD), eine Kollagenose, eine Herzinsuffizienz, eine Schädigung der Lunge durch Noxen oder eine Pneumonie hin. Außerdem gibt es rund 200 interstitielle Lungenerkrankungen. Umso anspruchsvoller ist es, die Erkrankung zu erkennen. Zwischen dem ersten Arztkontakt aufgrund von Beschwerden und der IPF-Diagnose können bis zu zwei Jahre vergehen.
Zu den Diagnoseverfahren gehören eine gründliche Anamnese und körperliche Untersuchung, Lungenfunktionstests wie die Spirometrie und die Diffusionskapazität (DLCO), Bildgebungsverfahren wie Röntgen und hochauflösender Computertomographie (HRCT) der Lunge und gegebenenfalls eine Lungengewebebiopsie.
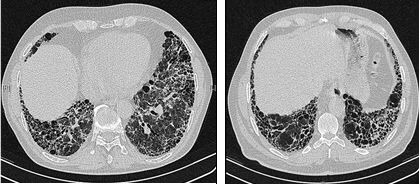
Das HRCT zeigt bei IPF Netzstrukturen mit Honigwabenmuster. Credit: IPFeditor/Wikimedia Commons.
Im Röntgen finden Ärzte mitunter strukturelle Veränderungen der Lunge. Gerade in frühen Stadien liefert die Methode kaum aussagekräftige Hinweise. Goldstandard ist das hochauflösende Thorax-CT (HRCT). Es zeigt bei Patienten ein sogenanntes UIP-Muster (Usual Interstitial Pneumonia) mit „Honigwaben“ (kleine, zystische Räumen, die von verdickten, vernarbten Lungenstrukturen umgeben sind), retikulären Verdichtungen und Traktionsbronchiektasen (mit pathologischen Erweiterungen der Bronchien). Milchglasartige Verdichtungen des Lungenparenchyms sind ebenfalls oft zu beobachten. Ohne solche Zeichen können eine Lungenbiopsie oder eine bronchoalveoläre Lavage durchgeführt werden.
Heilen lässt sich eine IPF rein pharmakologisch derzeit nicht. Alle Therapien zielen darauf ab, das Fortschreiten der Krankheit zu verlangsamen, Symptome zu lindern und die Lebensqualität zu verbessern. Sie sorgen auch für ein längeres Überleben der Patienten.
Leitlinien empfehlen als antifibrotische Pharmakotherapie derzeit nur die Wirkstoffe Pirfenidon und Nintedanib. Pirfenidon verringert die Synthese von Wachstumsfaktoren und von Kollagen-Vorstufen; Nintedanib hemmt als Tyrosinkinase-Inhibitor Fibroblasten. Die Wirkstoffe sollten symptomatischen Patienten ab dem Zeitpunkt der Diagnose verordnet werden, wobei Leitlinienautoren keine Kombination empfehlen. Sprechen Patienten darauf an, lohnt sich eine zeitlich unbefristete Behandlung – gegebenenfalls bis zur Lungentransplantation als einziger kurativer Möglichkeit.
Acetylcystein, Immunsuppressiva, Phosphodiesterasehemmer und andere Pharmaka empfehlen Autoren der deutschen Leitlinie nicht mehr. Große Hoffnungen setzen Forscher auf monoklonale Antikörper: Diese Biologicals richten sich gegen verschiedene Wachstumsfaktoren und Zytokine, von denen bekannt ist, dass sie bei der Proliferation, Aktivierung, Differenzierung oder dem Überleben von Fibroblasten eine Rolle spielen. Mehrere klinische Studien laufen, teils mit ernüchternden Ergebnissen. Simtuzumab hat beispielsweise Erwartungen zur Wirksamkeit nicht erfüllt.
Weitere Arbeitsgruppen fanden heraus, dass ein Algorithmus schon Jahre vor der klinischen Diagnose Patienten mit hohem Risiko für IPF identifizieren kann. Das bietet Chancen für eine deutlich frühere Intervention, verglichen mit dem Status quo.
Arzneimitteltherapien sollten mit Lebensstilinterventionen kombiniert werden, etwa mit einem sofortigem Rauchstopp, mit einer Normalisierung des Body-Mass-Index und mit individuell angepassten Bewegungsprogrammen. Patienten profitieren von umfassenden Programmen zur pneumologischen Rehabilitation mit Atemtherapie, Krankengymnastik, Ergotherapie, Entspannungstherapie, Ernährungsberatung, Tabakentwöhnung und weiteren Bausteinen.
Langzeit-Sauerstofftherapien bei niedrigem Sauerstoffpartialdruck sind ebenfalls sinnvoll. Und nicht zuletzt: Impfungen gemäß STIKO-Empfehlung verringern das Risiko von Infektionen und damit Progressionen der Erkrankung.
Bleibt als Fazit: Generell ist die Prognose schlecht. Das mittlere Überleben nach Diagnosestellung liegt bei drei bis fünf Jahren. Akute Exazerbationen stehen mit einer höheren Mortalität in Verbindung – die mittlere Lebenserwartung nach schweren Ereignissen liegt bei drei bis vier Monaten.
Bildquelle: Lucas Kapla, Unsplash