Wird ein Signalprotein durch Hemmstoffe blockiert, springt oft ein Schwesterprotein überregulierend ein. Das kann bei Krebsmedikamenten Probleme machen. Ein neuer Ansatz liefert eine mögliche Lösung.
Viele Krebsmedikamente blockieren in Krebszellen Signale, mit deren Hilfe sich entartete Zellen unkontrolliert vermehren und aus dem Gewebeverband herauslösen. So führt zum Beispiel die Blockade des Signalproteins FAK, einer Kinase, dazu, dass bestimmte Brustkrebszellen weniger beweglich werden und somit weniger stark metastasieren.
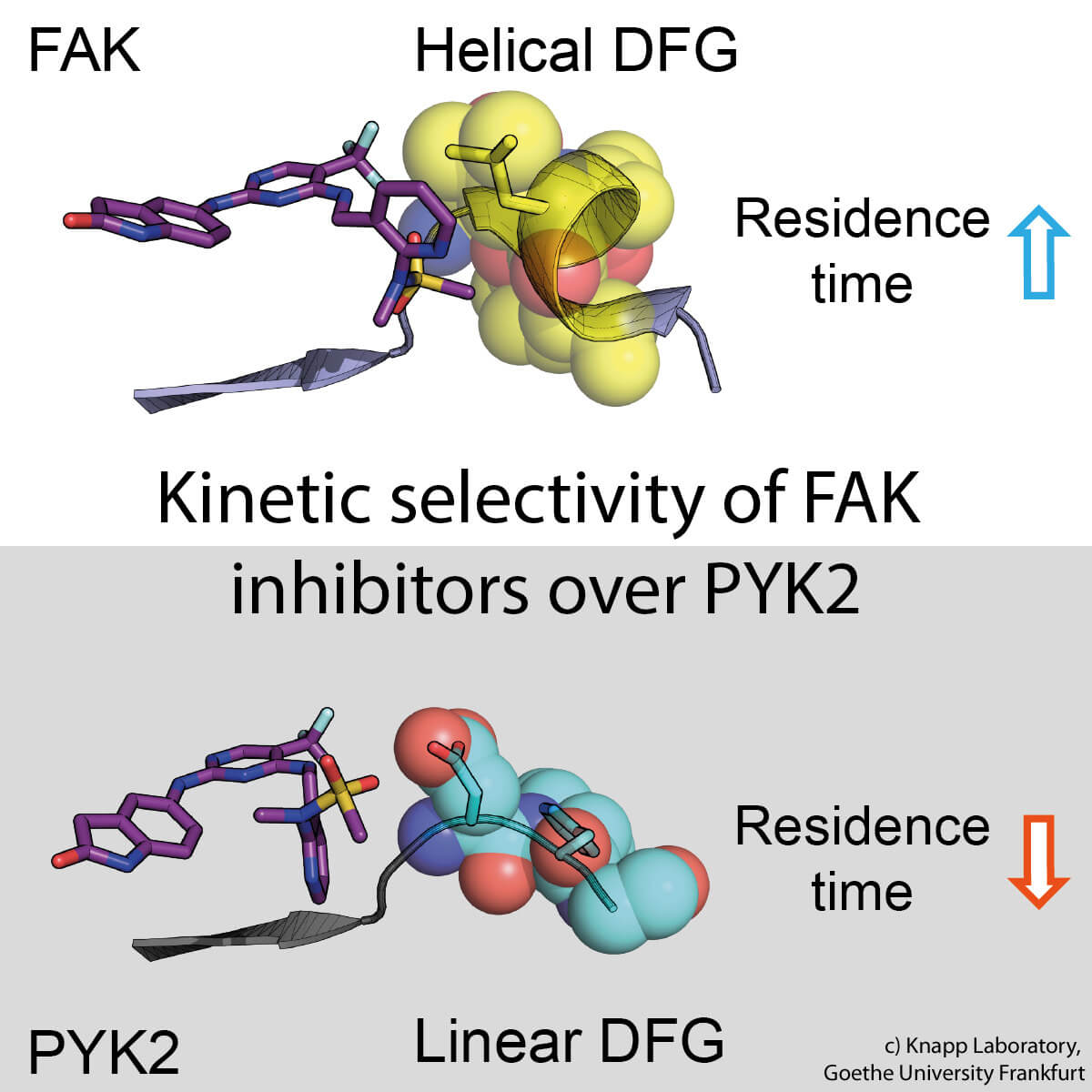
Das Problem: Wenn FAK durch einen Hemmstoff blockiert wird, wird das nahe verwandte Signalprotein PYK2 viel aktiver und übernimmt so einen Teil der Aufgaben von FAK. Ideal wäre daher ein Hemmstoff, der in gleicher Weise sowohl FAK wie auch PYK2 möglichst langanhaltend inhibiert.
Ein internationales Team um Pharmakochemiker Prof. Stefan Knapp von der Goethe-Universität hat eine Reihe eigens synthetisierter FAK-Hemmstoffe untersucht. Alle Hemmstoffe banden ungefähr gleich schnell an das FAK-Signalprotein. Sie unterschieden sich jedoch in der Dauer der Bindung: Der wirksamste Hemmstoff blieb am längsten mit dem FAK-Signalprotein verbunden.
In biochemischen und molekularbiologischen Analysen sowie Computersimulationen fand das Forschungsteam heraus, dass hierfür die Art der Wechselwirkung zwischen FAK-Signalprotein und Hemmstoff verantwortlich ist. Durch die Bindung des Wirkstoffs verändert das FAK-Signalprotein seine Form und bildet an einer der Kontaktstellen eine bestimmte, wasserabweisende Struktur aus. Diese induzierte FAK-Struktur bindet besonders gut an eine ebenfalls wasserabweisende Struktur des Hemmstoffs, vergleichbar einer innigen Umarmung.
Das Schwesterprotein PYK2 hingegen bleibt vergleichsweise steif, und obwohl der wirksamste FAK-Hemmstoff auch PYK2 blockierte, war sein Effekt hier deutlich schwächer. Den Forschern gelang es, das Bindungsverhalten der Inhibitoren in Computersimulationen zu modellieren und so eine Methode zu entwickeln, mit deren Hilfe sich künftig in der pharmazeutischen Forschung Wirkstoffkandidaten optimieren lassen.
Bildquelle: Goethe-Universität Frankfurt am Main
Knapp erklärt: „Weil wir jetzt die molekularen Mechanismen der Interaktion von potenten Hemmstoffen dieser zwei Kinasen besser verstanden haben, hoffe wir, künftig anhand von Computersimulationen die Verweildauer potenzieller Wirkstoffe besser vorhersagen zu können. Die Verweildauer von Wirkstoffen wurde bisher nur wenig beachtet. Diese Eigenschaft hat sich jedoch als wichtiger Parameter für die Entwicklung von effektiven Wirkstoffen entpuppt.“
Dieser Artikel basiert auf einer Pressemitteilung der Goethe-Universität Frankfurt am Main. Die Studie haben wir euch hier und im Text verlinkt.
Bildquelle: Morteza Yousefi, Unsplash