Für Orphan Drugs gelten spezielle Zulassungsverfahren, bestimmte Gebühren entfallen und auch ein Zusatznutzen muss nicht nachgewiesen werden. Ein neues Gesetz könnte das ändern. Was Patientenverbände besorgt, freut die Kassen.
Orphan Drugs sind Medikamente, die für seltene Erkrankungen entwickelt wurden und neu auf den Markt kommen. Kritiker beschweren sich schon seit geraumer Zeit über die dürftige Datenlage, die zu kleinen Patientengruppen und die hohen Kosten. Nun wurde offenbar beschlossen, einmal zu prüfen, ob an den Vorwürfen etwas dran ist. Ein neuer Entwurf zum Gesetz für mehr Sicherheit in der Arzneimittelversorgung (GSAV) vom 27.03.2019 sieht vor, dass der G-BA zukünftig mehr Einblicke in die Therapien erhält. Hersteller sowie Patientenverbände gehen dagegen auf die Barrikaden. Zurecht?
Orphan Drugs waren – wie die Namensgebung schon andeutet – einst so etwas wie die vergessenen Kinder der pharmazeutischen Industrie. Medikamente für Menschen mit seltenen Erkrankungen zu entwickeln lohnte sich kaum, denn man musste viel Geld in die Hand nehmen, um Wenigen zu helfen. Und da die Industrie kein Sozialstaat ist und sich die Entwicklung eben dieser Arzneimittel kaum rechnete, wurde eine Zeitlang zu wenig geforscht. Als selten gilt eine Krankheit in der Europäischen Union übrigens dann, wenn von durchschnittlich 2000 EU-Bürgern nicht mehr als einer von ihr betroffen ist.
Das änderte sich im Jahr 2000 mit der EU-Verordnung über Arzneimittel für seltene Leiden, nachdem die Politik in Zugzwang geriet. Seither gilt für Orphan Drugs ein anderes Zulassungsverfahren und die Hersteller erhalten für mindestens fünf und höchstens zehn Jahre eine garantierte Marktexklusivität. Auch der Nachweis für den Zusatznutzen, der seit 2010 für andere Arzneimittel gefordert wird, muss hier nicht erbracht werden. Manchmal entfallen sogar gewisse Gebühren bei der Zulassung, die ohne den Seltenheitsstatus anfallen würden.
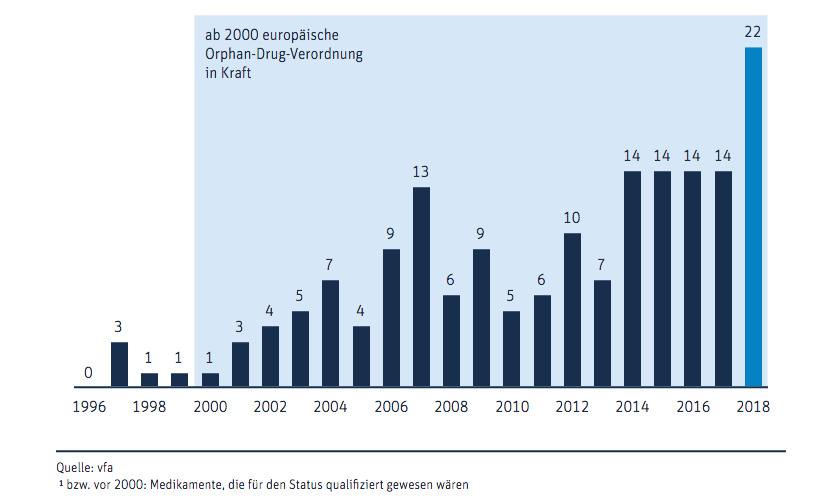
Derzeit sind 106 solcher Orphan Drugs auf dem Markt erhältlich, zusätzlich zu 52, die den Status zwar inzwischen zurückgegeben haben, aber den Patienten noch immer zur Verfügung stehen. Die Entwicklung von Orphan Drugs boomt.
Während der letzten fünf Jahre gehörten knapp über 30 Prozent der Neueinführungen auf dem Arzneimittelmarkt zu den sogenannten Waisen. Viele Krankenkassen waren darüber deutlich weniger erfreut, als die Patientenverbände von Menschen mit seltenen Erkrankungen. Kritiker monieren die angeblich zu dünne Datenlage, die zur Zulassung als Orphan Drug führe, und die hohen Kosten, die dadurch der Allgemeinheit entstünden. Das rief nun die Politik auf den Plan.
Im Gesetzesentwurf soll dem G-BA erlaubt werden, die anwendungsbegleitende Datenerhebung durch Ärzte und Krankenhäuser anzuordnen. Die Daten, für deren Erstellung die Hersteller verpflichtet werden können, sollen in ein Register eingepflegt werden. Sollte sich nach deren Auswertung kein Zusatznutzen durch die Orphan Drugs ergeben, könnten Abschläge auf den Erstattungsbetrag möglich sein. Welcher Art die Studien sind, die gefordert werden könnten (Fall-Kontroll-Studie, Anwendungsbeobachtung oder Registerstudie), soll dabei auch dem Ermessen des G-BA unterstehen.
Dr. Kai Joachimsen, Hauptgeschäftsführer des Bundesverbandes der Pharmazeutischen Industrie, sieht darin keine Bemühung um „wissenschaftlichen Erkenntnissgewinn“, sondern ist der Meinung, es gehe hier „eigentlich um Kostensenkungen, die Auswirkungen auf die Versorgung der Betroffenen haben werden.“ Er sieht „nicht absehbare (...) finanzielle Unsicherheiten“ auf die Hersteller und Entwickler von Orphan Drugs zukommen und befürchtet, dass „der Zugang zu Orphan Drugs (...) für Patienten erschwert“ wird.
Vfa-Hauptgeschäftsführerin Birgit Fischer sieht das ähnlich: „Ausgerechnet beim Thema seltene Erkrankungen biegt die Gesetzgebung in eine Sackgasse ein. Das trifft eine Patientengruppe, die die Unterstützung der Politik ganz besonders braucht.“
Die Vertreter von 120 Patientenorganisationen, die sich zum Netzwerk von und für Menschen mit (chronischen) seltenen Erkrankungen (ACHSE e.V.) zusammengeschlossen haben, befürchten ebenfalls Verschlechterungen für ihre Belange. In einer Stellungnahme heißt es: „Die ACHSE befürchtet negative Auswirkungen auf den Zugang zu den Medikamenten sowie auf die Forschung und Entwicklung, wenn eine solche neue Datenerhebung nicht im Vorfeld mit den verschiedenen Akteuren in Deutschland sowie den anderen HTA-Behörden in Europa abgestimmt wird.“
Die Vertreter der Krankenkassen sehen das völlig anders. Bereits im Jahr 2016 forderte der GKV-Spitzenverband genau das, was nun beschlossen werden soll. „Um dem G-BA bei seltenen Krankheiten eine sichere, am Patientennutzen orientierte Arzneimittelbewertung zu ermöglichen, fordert der GKV-Spitzenverband, dass er in begründeten Fällen auch bei Orphan Drugs das Nutzen- und Schadenspotenzial im Vergleich zum Therapiestandard vollständig prüfen dürfen soll.“
G-BA Vorsitzender Josef Hecken, der in den letzten Jahren besonders häufig die hochpreisigen Arzneimittel kritisierte, die keinen Zusatznutzen nachweisen mussten, sagte dazu gegenüber dem Ärzteblatt: „Wir wollen ja nicht autistisch nur in Deutschland Daten erheben (...). Ich befürchte auch in keiner Weise, dass ein Hersteller den deutschen Markt verlässt, wenn es Anwendungsbeobachtungen gibt.“
Man darf gespannt bleiben, ob er mit dieser Aussage Recht behalten wird.
Artikel von Eva Bahn
Bildquelle: per Corell, Flickr