Patienten, aber auch Herstellern, kann es nicht schnell genug gehen, bis ein neuer Arzneistoff endlich zugelassen wird. Jetzt sägen Gesundheitspolitiker am Stuhl der Ethikkommission, aber auch an klinischen Studien. Dabei gibt es längst Alternativen.
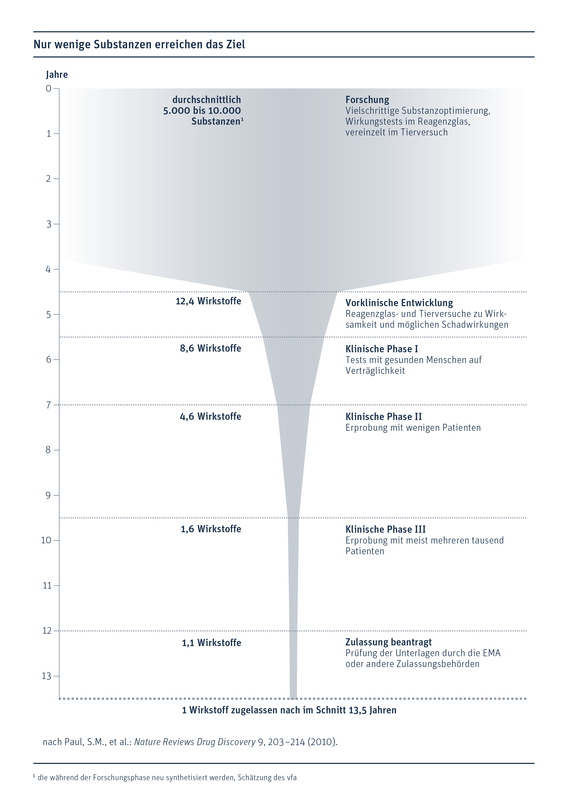
Vom ersten Experiment im Labor bis zur Zulassung des neuen Präparats vergehen im Schnitt mehr als 13 Jahre, berichtet der Verband forschender Arzneimittelhersteller (VFA). Davon entfallen vier bis fünf Jahre auf die Grundlagenforschung, um geeignete Wirkstoffe zu identifizierten und erste Tests im Tierexperiment durchzuführen. Weitere fünf bis sechs Jahre gehen auf das Konto klinischer Studien: ein Kriterium, das mit zu sicheren Präparaten beiträgt. Dieser zeitliche Vorlauf gefällt nicht allen Herstellern.
Professor Jörg Hasford. Quelle: Arbeitskreis medizinischer Ethik-Kommissionen in der Bundesrepublik Deutschland Jetzt tritt Bundesgesundheitsminister Hermann Gröhe (CDU) auf das Gaspedal. Mit seinem geplanten Entwurf des „Vierten Gesetzes zur Änderung arzneimittelrechtlicher und anderer Vorschriften“ will er die Macht von Ethikkommissionen deutlich einschränken. Momentan entscheiden diese, ob klinische Studien überhaupt zulässig sind. Anfang des Jahres war in Frankreich ein Freiwilliger im Rahmen einer klinischen Studie ums Leben gekommen. Damals sei die Medikamentenstudie so konzipiert gewesen, dass Schnelligkeit vor Patientenwohl ging, erklärte Professor Jörg Hasford, Vorsitzender des Arbeitskreises medizinischer Ethik-Kommissionen in der Bundesrepublik Deutschland, bei einer Anhörung im Bundestag. Experten sehen auch Deutschland in Gefahr: Eigenständig handelnde Ethikkommissionen könnten der Vergangenheit angehören, falls sich Gröhe durchsetzt. Sein Entwurf sieht vor, dass sich Kommissionen beim Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) registrieren müssen, inklusive Möglichkeit des Widerrufs. Gleichzeitig wären BfArM-Experten nicht mehr an das Votum von unabhängigen Experten gebunden. Auch eine Bundes-Ethikkommission direkt am BfArM scheint denkbar zu sein. Hasford ist besorgt: „Bei 97 Prozent der Anträge auf klinische Studien sehen wir teils erheblichen Nachbesserungsbedarf.“ Oft seien Nebenwirkungen oder Angaben zu sonstigen Behandlungsmöglichkeiten außerhalb einer Studie mangelhaft und für Laien nicht verständlich dargestellt. „Wir müssen aber gewährleisten, dass die Patienten wissen, auf was sie sich einlassen“, sagte der Experte.
Ähnliche Gefahren drohen von zentraler Stelle: Auch die europäische Arzneimittelbehörde EMA plant Lockerung der Zulassungsvorschriften für neue Arzneimittel, um Präparate schneller in den Markt zu bringen. EMA-Experte Hans-Georg Eichler hat zusammen mit Kollegen zwei Strategiepapiere veröffentlicht: „From adaptive licensing to adaptive pathways: Delivering a flexible life-span approach to bring new drugs to patients“ sowie „Adaptive Licensing: Taking the Next Step in the Evolution of Drug Approval“. Die von ihm beschriebene adaptive Zulassung sieht vor, dass künftig kleine Patientenzahlen ausreichen, um eine „bedingte Zulassung“ zu erhalten. Hersteller haben die Möglichkeit, weitere Daten nachzureichen, sobald sich ihr Präparat auf dem Markt befindet. Eichler und Kollegen argumentieren mit der Chance, Patienten schneller zu versorgen als bisher. In den Artikeln ist aber auch von „früheren Einkünften und weniger teuren und kürzeren klinischen Studien“ die Rede. Pilotprojekte laufen wenig beachtet bereits seit 2004. Quelle: VFA
MEZIS („Mein Essen zahl ich selbst“) befürchtet nicht nur Abstriche bei der Sicherheit und Gefahren durch neue, wenig erprobte Medikamente. Der industriekritische Verband sieht auch gravierende Änderungen bei der Beweislastumkehr auf Europa zukommen. Statt Hersteller zu verpflichten, hochwertige Daten vorab einzureichen, müssen vielleicht schon bald Versicherte und Health Professionals Gefahren nachweisen, damit ein Präparat wieder vom Markt genommen wird. Auch die Veröffentlichungen über adaptive Zulassungen stehen auf tönernen Füßen. Zahlreiche Autoren arbeiten für die pharmazeutische Industrie. Genannt werden unter anderem Novartis Vaccines & Diagnostics, AstraZeneca, Bristol-Myers Squibb, Johnson & Johnson oder Pfizer. „Die Allianz aus Zulassungsbehörden und Unternehmen erscheint besonders bedenklich, wenn Deregulierungen von derartiger Tragweite vorangetrieben werden“, schreibt MEZIS-Geschäftsführerin Dr. Christiane Fischer. „Wie die EMA in dieser Konstellation ihre Unabhängigkeit als eine der ranghöchsten Hüterinnen der öffentlichen Gesundheit in Europa zu wahren gedenkt, ist nicht bekannt.“ Unklar bleibt auch, in welchen Fällen das adaptive Konzept greifen soll. Während die EMA auf ihrer Website „areas of high medical need“ („Bereiche mit erheblichem medizinischem Bedarf“) nennt, sieht Hans-Georg Eichler neue Zulassungsverfahren als „Ersatz für das derzeitige Zulassungs- und Autorisierungsmodell“.
Dass es hier tatsächlich nicht um die Versorgung von Patienten geht, zeigen derzeit mögliche Ausnahmeregelungen. Stehen tatsächlich keine Arzneimittel zur Verfügung, können Hersteller Härtefallprogramme („Compassionate Use“) für Patienten aufsetzen. Eine Übersicht für Deutschland inklusive Entscheidungsgrundlagen veröffentlicht das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM). Individuelle Heilversuche oder Off-Label-Verordnungen ergänzen das Spektrum an Möglichkeiten. Fabian Schubach von MEZIS kritisiert beim europäischen Vorstoß deshalb den „zu erwartende ökonomische Nutzen für Pharmafirmen – auf Kosten der Sicherheit von Patienten“.