Sollen Studiendaten frei zugänglich sein oder zumindest in Teilen unter Verschluss bleiben? Darüber streiten Behörden und Hersteller schon seit Langem. Gerichtliche Auseinandersetzungen tragen kaum zur Klärung bei.
Premiere vor dem Kadi: Zum ersten Mal hat eine Privatperson in Deutschland gegen einen pharmazeutischen Hersteller geklagt. Roland Holtz, selbst zu früheren Zeiten in der Arzneimittelbranche tätig, zweifelt an Studiendaten zu Dabigatran. In der Anklageschrift schreibt er, Boehringer Ingelheim habe die europäische Arzneimittelzulassungsbehörde EMA (European Medicines Agency) bei deren Anfrage im März 2012 „durch Verschweigen wesentlicher sicherheitsrelevanter Risiken, nämlich der Variabilität des Plasmaspiegels, im Umgang mit dem Präparat Pradaxa® getäuscht“.
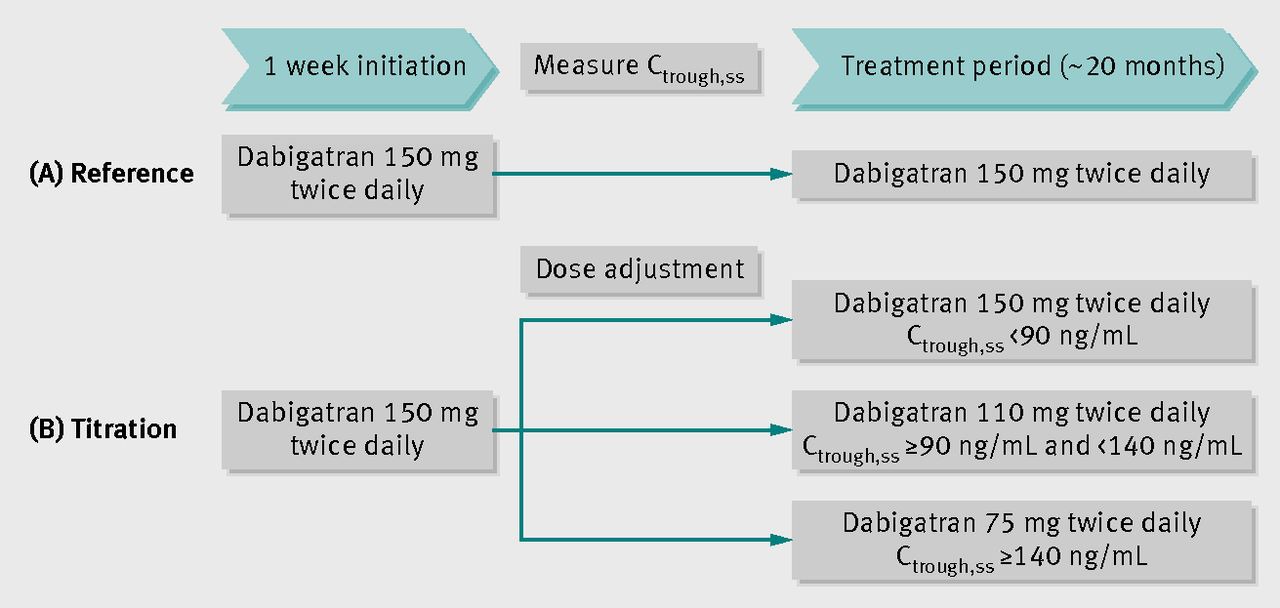
Der Kritiker argumentiert vor allem mit internen Daten, die Boehringer Ingelheim EMA-Experten verschwiegen habe. Deshalb sei auf regelmäßige Bluttests wie bei Phenprocoumon (Marcumar®) verzichtet worden – ein wesentliches Argument im Marketing. Holtz zufolge habe das British Medical Journal (BMJ) mehrfach Dokumente unter Berufung auf das Informationsfreiheitsgesetz direkt von der EMA erhalten. „Dabigatran: how the drug company withheld important analyses“, notierte BMJ-Investigation Editor Deborah Cohen bereits vor zwei Jahren. Deborah Cohen schreibt im BMJ, Boehringer Ingelheim habe ursprünglich sehr wohl ein Titrationsschema bei bei Dabigatran vorgesehen, um Blutungsrisiken zu minimieren. Grafik: BMJ In der Anklageschrift heißt es, die Angaben über Plasmavariabilitäten der Wirkspiegel seien „unzutreffend und deutlich zu niedrig“. Juristen von Boehringer Ingelheim präsentierten ihrerseits eine Verteidigungsschrift, um alle Vorwürfe mit Verweis auf Zulassungsdaten zu widerlegen. Die Staatsanwaltschaft Mainz sah schließlich keinen Anfangsverdacht und wird auch kein Ermittlungsverfahren initiieren. Es lägen weder Verstöße gegen das Arzneimittelgesetz noch gegen das Verbot des Inverkehrbringens von bedenklichen Arzneimitteln vor, heißt es zur Begründung. Ein schaler Nachgeschmack bleibt, und Wissenschaftler fordern umso lauter Transparenz bei klinischen Studien.
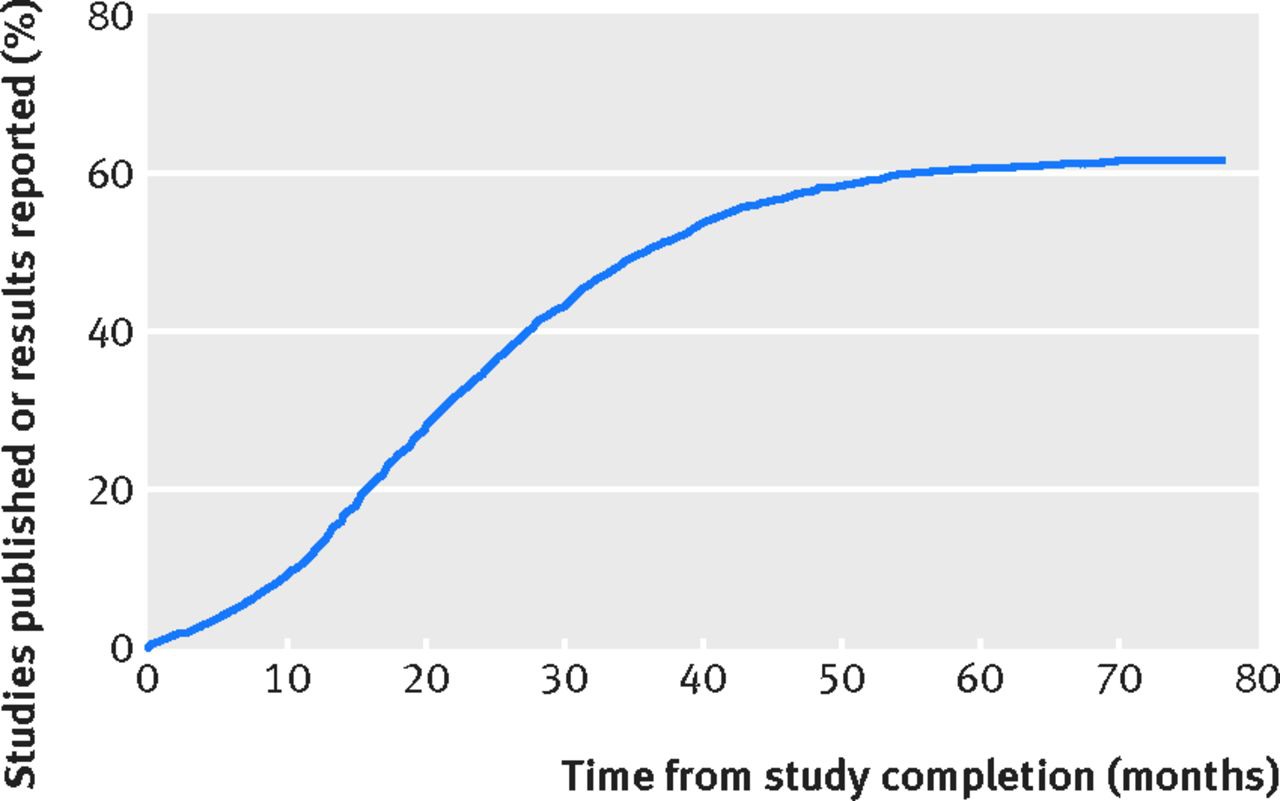
Dazu ein paar Zahlen. Harlan M. Krumholz aus New Haven (Connecticut) ging der Frage nach, wie oft Daten aus klinischen Studien tatsächlich veröffentlicht werden. Zusammen mit Kollegen identifizierte er 4.347 Arbeiten aus 51 akademischen Zentren. Darunter waren 1.005 Studien mit mehr als 100 Patienten. Es gab 1.216 doppelt verblindete Studien, und 2.169 Phase-II- bis Phase-IV-Studien. Innerhalb von 24 Monaten wurden nur zu jedem dritten Projekt Informationen publiziert. Nach 36 Monaten waren es rund 60 Prozent. Quelle: BMJ Oft stecken Kooperationspartner hinter der fehlenden Transparenz. Deutschland ist hier keine Ausnahme, wie folgendes Verfahren zeigt: Mitte 2015 urteilte das Oberverwaltungsgericht Münster, dass die Uni Köln keine Details zur Kooperation mit der Bayer AG bekanntgeben muss (Aktenzeichen: 15 A 97/13). Andere Knebelverträge verbieten Forschern auch, Resultate in Zeitschriften zu veröffentlichen oder auf Kongressen zu präsentieren.
Europäischen Politikern ist die Kontroverse nicht neu. Jede Studie wird automatisch mit der Antragstellung in der EudraCT-Datenbank (European Union Drug Regulating Authorities Clinical Trials) registriert. Einträge für Phase-I-Studien stehen nur Behörden oder Ethik-Kommissionen zur Verfügung. Nach massiver Kritik gelangen Ergebnisse abgeschlossener Studien in Teilen nach außen. Das EU Clinical Trials Register enthält Informationen über pädiatrische klinische Studien gemäß „Paediatric Investigation Plan“ und Studien der Phasen II bis IV mit Erwachsenen. Ab 2018 greift die Clinical Trials Regulation, bekannt als EU-Verordnung 536/2014. Ziel ist, eine zentrale Plattform zu schaffen, um Studien komplett abzubilden. „Damit bei den klinischen Prüfungen ein ausreichendes Maß an Transparenz besteht, sollten in dieser EU-Datenbank alle über das EU-Portal übermittelten relevanten Informationen zu klinischen Prüfungen erfasst werden“, heißt es zur Begründung. Der Bogen spannt sich von der Antragstellung, Genehmigung und Registrierung über die Studienbegleitung bis hin zur Veröffentlichung der Ergebnisse. Zur Planung heißt es weiter: „Die EU-Datenbank sollte öffentlich zugänglich sein, und die Daten sollten in einem Format präsentiert werden, das die Suche erleichtert [...].“ Doch der Teufel steckt im Detail. „Für die Zwecke dieser Verordnung sollten die in einem Studienabschlussbericht enthaltenen Daten grundsätzlich nicht mehr als vertrauliche geschäftliche Informationen gelten, sobald eine Zulassung erteilt wurde, das Verfahren zur Erteilung einer Zulassung abgeschlossen ist oder der Antrag auf Zulassung zurückgezogen wurde“, schreiben EU-Politiker.
Dieser Passus stößt teilweise auf Widerstand. Beim Pharmadialog meldeten sich Hersteller mit deutlicher Kritik zu Worte. Sie forderten von Bundesgesundheitsminister Hermann Gröhe (CDU), Sorge zu tragen, dass der Schutz von Betriebs- und Geschäftsgeheimnissen ausreichend berücksichtigt wird. Beate Wieseler, Leiterin des Ressorts Arzneimittelbewertung am Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG) hatte Anfang 2015 gefordert, die Definition von Geschäftsgeheimnissen zu präzisieren, damit Behörden Zugriff auf alle relevanten Informationen erhalten.