Implantate aus dem 3D-Drucker setzen sich in der Medizin immer mehr durch. Aber wie lässt sich ihre Qualität überwachen? Die amerikanische FDA hat jetzt eine neue Richtlinie vorgestellt. Das BfArM hingegen tut das, was es am besten kann: Es wartet erstmal ab.
Die 3D-Druck-Technologie macht immer größere Sprünge – auch beim Einsatz in der Medizin. Dafür gibt es gleich mehrere spektakuläre Beispiele: So konnten 2014 Ärzte im chinesischen Xijing einem Mann helfen, dessen Schädeldecke bei einem Sturz aus dem dritten Stock zerstört worden war. Die Chirurgen entfernten die zersplitterten Knochenanteile und fertigten dann mithilfe eines 3D-Druckers Ersatz – eine passgenaue Netzstruktur aus Titan. Im niederländischen Utrecht setzten Ärzte ebenfalls schon vor zwei Jahren einer Patientin mit einer seltenen Knocherkrankung eine Schädeldecke aus Plastik ein. Mit dem 3D-Drucker ließ sich Ersatz produzieren, der genau der richtigen, natürlichen Kopfform der Frau entsprach. Bei Einzelfällen wird es nicht bleiben: Zu Beginn diesen Jahres erteilte die amerikanische Arzneimittelbehörde FDA die Zulassung von Titan-Schädelplatten aus dem 3D-Drucker. Und schon vor fünf Jahren genehmigte sie per 3D-Druck gefertigte Knie-Implantate.
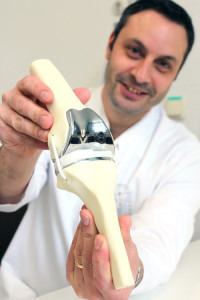
Die Vorteile der neuen Technologie liegen auf der Hand. Anders als Einheitsware können 3D-Implantate individuell gefertigt werden. Mit dem Computer wird vorab exakt berechnet, was für ein Ersatzteil benötigt wird. Selbst komplexe Formen und Strukturen lassen sich so passgenau drucken. Zudem ist die Methode oft kostengünstiger als bisher gängige Technologien. Dr. I. Chatziandreou mit einer Knieprothese aus dem 3D-Drucker © Klinikum Dortmund Aber wie sicher ist das Verfahren? Gerade die individuelle Fertigung kann Qualitätskontrollen erschweren. Schließlich kann nicht jedes Unikat einzeln von den Behörden genehmigt werden. Die amerikanische Arzneimittelbehörde FDA hat nun einen Richtlinienentwurf verfasst. Darin gibt sie Empfehlungen, die sich auf die Produktion und Qualitätstests von Medizinprodukten aus dem 3D-Drucker beziehen. Bereits 2014 hatte die FDA einen öffentlichen Workshop zum Thema veranstaltet, bei dem Experten und Hersteller über Anforderungen an die Produktion von 3D- Medizinprodukten diskutierten. Der Austausch mit den Herstellern sollte für mehr Sicherheit sorgen und ihnen den Weg zur Zulassung ihrer Produkte ebnen, um Innovationen zu fördern.
Auch in Deutschland sind Medizinprodukte aus dem 3D-Drucker schon im Einsatz. So werden Patienten bereits in mehreren Kliniken Knieprothesen aus dem Drucker eingesetzt. Vom Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) gibt es aber noch keine Richtlinie für das neue Verfahren. Man hat nicht einmal einen Überblick dazu, in welchem Umfang Medizinprodukte aus dem 3D-Drucker bereits eingesetzt werden. Denn anders als die amerikanische FDA ist das BfArM zwar für die Zulassung von Arzneimitteln, nicht aber für die Zulassung von Medizinprodukten zuständig. Hersteller von Implantaten müssen stattdessen bei einer externen Prüfstelle eine Zertifizierung erwerben. „Die gesetzliche Aufgabe des BfArM ist bei Medizinprodukten vor allem die Risikobewertung‟, sagt der Sprecher der Behörde, Maik Pommer. Man werde dann tätig, wenn zu Produkten, die bereits am Markt sind, Vorkommnisse gemeldet werden. Empfehlungen spricht das BfArm erst dann aus, wenn der Verdacht besteht, dass ein Medizinprodukt fehlerhaft oder nicht sicher für die Patienten sei. Zwar informiere die Behörde auch vorab zu bestimmten Themen: „Etwa dann, wenn mit Blick auf den Patientenschutz ein besonders hoher Informationsbedarf besteht‟, sagt Pommer, und verweist auf ein aktuelles Symposium des BfArM, bei dem es um Health Apps geht. Beim 3D-Druck sehe man derzeit noch nicht den Bedarf. Hersteller solcher Medizinprodukte könnten sich aber vom BfArM jederzeit kostenpflichtig beraten lassen, so Pommer.
Insgesamt sind die Befugnisse des BfArM gegenüber Behörden wie der FDA bei Medizinprodukten deutlich eingeschränkt. Dass das problematisch sein kann, zeigte sich in der Vergangenheit mehrfach. So wurde 2010 bekannt, dass der französische Hersteller PIP mit Industriesilikon gefüllte Brustimplantate in Umlauf gebracht hatte, die noch dazu häufig undicht wurden – aber vom TÜV-Rheinland zertifiziert waren. Während die französische Medizinproduktebehörde die Anwendung direkt untersagen konnte, konnte das BfArM lediglich empfehlen, die Produkte nicht mehr zu implantieren. Zu einem Verbot war sie nicht ermächtigt. Zum Vergleich: Die FDA hatte nach einem früheren Skandal schon fast 20 Jahre zuvor klinische Daten zur Sicherheit von Silikon-Brustimplantaten von sämtlichen Herstellern eingefordert. Als diese der Aufforderung nicht nachkamen, entzog die FDA ihnen die Zulassung und genehmigte erst Jahre später unter strengeren Auflagen solche Produkte. Der GKV-Spitzenverband wies schon 2012 eindringlich auf diese Problematik hin. Das BfArM müsse „bei erkannten Medizinprodukt-Anwendungsrisiken Maßnahmen ergreifen können, die über eine bloße Abgabe von Empfehlungen und Veröffentlichung derselben hinausgehen‟, forderte der Verband in einer Stellungnahme.
Auch bei Hüftimplantaten vom Typ „ASR‟ der Firma DePuy häuften sich über einen längeren Zeitraum hinweg negative Berichte. Patienten klagten über Schmerzen, in ihrem Blut fanden sich Metallionen aus dem Abrieb der Implantate. Pommer zufolge empfahl das BfArM schon 2008 dem Hersteller und der zuständigen Landesbehörde, das Produkt vom Markt zu nehmen. Bei einem Hustensaft wäre das möglich gewesen. Weil es sich aber nicht um ein Arzneimittel, sondern um ein Medizinprodukt handelte, war das BfArM selbst dazu nicht befugt. Und das zuständige saarländische „Landesamt für Umwelt- und Arbeitsschutz‟ kam der BfArM-Empfehlung nicht nach. Weitere Patienten nahmen Schaden, bis sich der Hersteller 2010 selbst entschloss, die Implatate vom Markt zu nehmen. In den nächsten Jahren ist eine Schwemme an neuartigen Medizinprodukten zu erwarten. Implantate aus dem 3D-Drucker sind dafür nur ein Beispiel, auch die erwähnten Health Apps zählen dazu. Es wäre effektiver und im Interesse der Patienten, deren Qualitäts-Überwachung komplett in die Hände einer Behörde zu legen. Diese muss die Möglichkeiten haben, präventiv zu handeln – und nicht erst dann, wenn es für viele zu spät ist.