Bereits seit Februar 2017 ist das erste Biosimilar (Rituximab) gegen Krebs in der EU zugelassen. Weitere Biosimilars folgten oder sind aktuell im Zulassungsverfahren. Trotzdem sind viele Onkologen noch zurückhaltend. Zu Unrecht, denn Biosimilars sind so gut wie das Referenzprodukt. Sie verfügen über Vorteile, die die Versorgung der Patienten verbessert.
Das Wichtigste zuerst: Biosimilars sind keine Generika. Bei diesen wird der molekular kleine Wirkstoff chemisch synthetisiert und stimmt daher mit dem im Originalpräparat überein. Ein Biosimilar ist ein Analogon zu einem komplexen biologischen Arzneimittel, welches bereits in der EU zugelassen wurde, das sogenannte Referenzarzneimittel. Das Biosimilar ist der Referenzsubstanz soweit ähnlich wie die verschiedenen Chargen der Referenzsubstanz zueinander ähnlich sind.
Nachweis der Ähnlichkeit der physikochemischen und biologischen Eigenschaften
Alle gentechnisch hergestellten Arzneimittel müssen in einem zentralisierten Verfahren über die europäische Arzneimittelbehörde (European Medicines Agency; EMA) zugelassen werden. Hier werden die Qualität, die Wirksamkeit, die Sicherheit und die Verträglichkeit der Präparate nach festen Regeln überprüft. Damit ein Biosimilar zugelassen werden kann, muss die Ähnlichkeit mit dem Referenzarzneimittel in umfangreichen pharmakologischen, toxikologischen und klinischen Studien nachgewiesen werden. Dieses Verfahren ist wesentlich aufwendiger und kostspieliger als das Zulassungsverfahren für klassische Generika.
Die Zulassung durch die EMA ist deshalb ein „Gütesiegel“, auf das sich Ärzte und Patienten verlassen können. Auch der Leitfaden der Arzneimittelkommission der deutschen Ärzteschaft (AkdÄ) vom August 2017 sieht die Anwendung von Biosimilars hinsichtlich Wirksamkeit, Qualität und Unbedenklichkeit als ausreichend erwiesen an. Ebenso das Paul-Ehrlich-Institut, die DGHO und die ESMO. Sie bewerten Biosimilars als eigenständige Präparate, die sowohl hinsichtlich der Wirksamkeit wie auch der Sicherheit den Erstanbieterpräparaten ebenbürtig sind.
Bei nicht vortherapierten Patienten macht es damit faktisch keinen Unterschied, ob die Referenzarznei oder ein Biosimilar eingesetzt wird. Das belegen gleichfalls die bisher vorliegenden Ergebnisse randomisierter Studien aus der Rheumatologie und der Onkologie. Sie zeigen keine signifikanten Unterschiede bezüglich Pharmakokinetik, Wirksamkeit und Sicherheit. Aus Sicht der AkdÄ und des Paul-Ehrlich-Instituts spricht deshalb auch bei vortherapierten Patienten nichts gegen eine Umstellung vom Referenzarzneimittel auf ein Biosimilar.
Chancen für Ärzte, Patienten und Gesundheitssysteme
Durch die Verordnung von Biosimilars können im Gesundheitswesen erhebliche Einsparungen realisiert werden. Bisherige Erfahrungen zeigen, dass die Kosten um 20 bis 30 Prozent niedriger ausfallen als bei den Referenzarzneimitteln.
Das bedeutet, dass damit für Patienten flächendeckend und nachhaltig weitere substanzielle Ressourcen für eine bestmögliche Versorgung zur Verfügung stehen. Sie erhalten einen verbesserten Zugang zu einer qualitativ hochwertigen Medikation. Gleichzeitig kann durch die Einsparungen bei Biosimilars auf dieser Basis die Versorgung der Patienten mit innovativen, hochpreisigen Arzneimitteln sichergestellt werden.
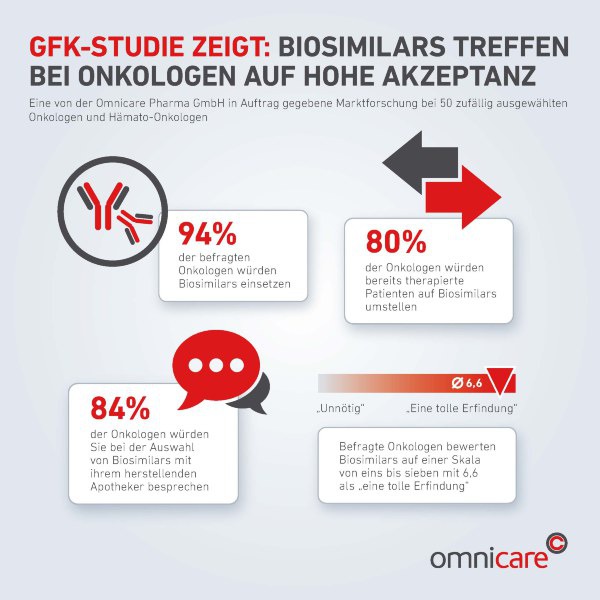
Marktforschung zeigt hohe Akzeptanz bei Onkologen
Einer Umfrage des Marktforschungsinstituts GfK zufolge sind inzwischen 94 Prozent der Onkologen bereit, Biosimilars einzusetzen. 80 Prozent würden auch bereits therapierte Patienten auf Biosimilars umstellen. Apotheker sind dabei wichtige Partner des Onkologen. 84 Prozent würden sich bei der Auswahl von Biosimilars mit ihrem Zytostatika-herstellenden Apotheker besprechen und dann die Entscheidung fällen.
Biosimilars sind inzwischen ein wichtiges Element der onkologischen Versorgung. Sie haben sich als wirtschaftlich sinnvolle und therapeutisch gleichwertige Option bewährt und kommen immer schneller in der ambulanten Versorgung an.